0. Beyond AlphaFold2: The Impact of Protein Structure Prediction on Small Molecule Drug Discovery
1. Introduction: The Evolving Landscape of Protein Structure Prediction and Drug Discovery
Proteins are central to all biological processes, with their intricate three-dimensional structures dictating their diverse functions, ranging from material transport and energy conversion to enzymatic catalysis . Consequently, understanding protein structure is paramount for elucidating disease mechanisms and for the rational design of therapeutic interventions. Aberrations in protein folding and structure are frequently linked to various pathological conditions, including neurodegenerative diseases and metabolic disorders . Given that the majority of small molecule drug targets are proteins, accurate structural information is indispensable for structure-based drug design (SBDD), which aims to develop novel and more effective therapeutics .
| Method | Description | Limitations |
|---|---|---|
| X-ray Crystallography | Determines protein structures from crystalline samples. | Requires stable protein crystals, time-consuming, expensive. |
| NMR Spectroscopy | Determines protein structures in solution. | Requires large sample quantities, challenges with dynamic/membrane proteins. |
| Cryo-Electron Microscopy (Cryo-EM) | Determines structures using electron beams on frozen samples. | Dependent on sample quality, challenges with dynamic/membrane proteins. |
| Overall Limitations | Significant time & cost, need for stable crystals/large samples, dynamic/membrane proteins challenging, disparity between sequence & structure (Levinthal paradox). |
Historically, experimental methodologies such as X-ray crystallography, Nuclear Magnetic Resonance (NMR) spectroscopy, and, more recently, cryo-electron microscopy (Cryo-EM), have been the primary means of determining protein structures . While these techniques have provided invaluable insights and populated databases like the Protein Data Bank (PDB) with static snapshots of protein conformations, they are often constrained by significant limitations. These include considerable time and cost requirements, the need for stable protein crystals or large sample quantities, and particular challenges with dynamic or membrane-bound proteins . These limitations have led to a substantial and growing disparity between the number of known protein sequences and experimentally determined structures, a problem encapsulated by the Levinthal paradox .
To address this gap, computational approaches emerged as crucial complementary tools. Early computational methods, such as homology modeling, offered a practical alternative when experimental structures were unavailable, relying on sequence similarity to predict structures based on known homologous proteins . The evolution of computational power, including advancements in multi-core processors and GPUs, further propelled the field, enabling more complex simulations and quantitative structure-activity relationship (QSAR) studies crucial for drug design . Computer-aided drug design (CADD) techniques specifically facilitated the characterization of ligand-binding mechanisms and active site identification, thereby enhancing the efficiency and success rates of the drug discovery process by reducing both time and financial investments .
The availability of accurate protein structures profoundly impacts drug discovery by enabling the precise identification of drug binding pockets, facilitating the rational design of small molecules with optimized fit, affinity, and selectivity, and ultimately minimizing off-target effects and toxicity . Despite these advancements, the inherent limitations of both experimental and early computational methods posed a continuous challenge to the efficiency and success of drug discovery.
The landscape of protein structure prediction was fundamentally reshaped with the advent of AlphaFold2 (AF2) in 2020. Developed by DeepMind, this deep learning-based AI model represents a transformative breakthrough, effectively solving the decades-old challenge of accurately predicting atomic-level protein structures from amino acid sequences alone . AlphaFold2 predicts protein structures with remarkable accuracy, often comparable to experimental methods, and at unprecedented speeds, with human protein 3D structures reported to be around 98.5% accurate . This breakthrough has been heralded as a major scientific advancement, significantly democratizing access to structural data and accelerating insights into protein function and interactions .
| Previous Methods | Limitations | AlphaFold2 Advantages |
|---|---|---|
| Experimental | Laborious, time-consuming, expensive, sample dependent. | N/A (AI-based) |
| Early Computational | Accuracy constraints, applicability limitations. | Leverages novel attention-based neural networks. |
| Relied on homologous structures or sequence similarity. | Directly predicts atomic coordinates with near-atomic precision. | |
| Trained on extensive PDB & UniProt databases. | ||
| Impact | Democratized access to structural data, accelerated research. |
AlphaFold2's "paradigm shift" status is attributed to its ability to overcome the limitations of prior methods. Unlike laborious and time-consuming experimental techniques or early computational methods that faced accuracy and applicability constraints, AlphaFold2 leverages novel attention-based neural network architectures and extensive training on databases like the PDB and UniProt to directly predict atomic coordinates with near-atomic precision . The release of AlphaFold2's source code and a vast dataset of over 350,000 protein structures, including the entire human proteome, has further revolutionized the field by making over 200 million protein structures globally accessible .
This paradigm shift has had an immediate impact on traditional drug discovery workflows. By providing highly accurate target protein structures, AlphaFold2 significantly accelerates SBDD, enabling the design of targeted and effective small molecule drugs, particularly for novel protein targets lacking experimentally determined structures . Examples include its integration into AI-driven drug discovery platforms to identify novel molecules, such as a CDK20 small molecule inhibitor, for targets previously inaccessible due to structural data scarcity . Furthermore, its capability to predict protein-protein interactions aids in understanding disease mechanisms and developing targeted therapies, addressing critical bottlenecks in the drug discovery process . While AlphaFold2 primarily offers static predictions of stable protein conformations and does not inherently capture the full range of protein dynamics crucial for certain drug interactions, its high accuracy (with pLDDT scores above 80 typically sufficient for SBDD) significantly expedites the initial phases of drug design .
1.1 Significance of Protein Structure in Drug Discovery
Proteins are fundamental to biological processes, executing vital activities such as material transport, energy conversion, and catalytic reactions. Their three-dimensional structure is inextricably linked to their function, making structural elucidation critical for understanding disease mechanisms and rational drug design . Deviations from native protein structures are implicated in various pathologies, including Alzheimer's, Parkinson's, and cystic fibrosis, underscoring the importance of understanding protein folding for therapeutic development . The majority of therapeutic targets for small molecule drugs are proteins, emphasizing the critical role of accurate protein structures in structure-based drug design (SBDD) to rationally develop novel therapeutics with improved outcomes .
Historically, protein structures were primarily determined using experimental methods such as X-ray crystallography and Nuclear Magnetic Resonance (NMR) spectroscopy . While these techniques have significantly contributed to the Protein Data Bank (PDB), providing essential static snapshots of proteins, they inherently possess limitations. Experimental methods are often time-consuming, expensive, and complex, requiring stable protein crystals for X-ray crystallography or significant sample quantity for NMR, which is particularly challenging for dynamic or membrane-bound proteins . This results in a substantial disparity between the rapidly growing number of known protein sequences and the comparatively slow rate of experimentally determined structures, a phenomenon highlighted by the Levinthal paradox . Even cryo-electron microscopy (Cryo-EM), while capable of determining structures for unstable proteins, remains dependent on sample quality .
To bridge this gap, computational approaches emerged as indispensable alternatives for acquiring structural information. Early computational methods, such as homology modeling, offered a viable path when experimental structures were unavailable . This technique relies on the existence of a known homologous protein structure as a template, inferring the target protein's structure based on sequence similarity. However, its accuracy was inherently limited by the availability and quality of suitable template structures . Beyond simpler techniques, the evolution of computational methods in drug discovery, prior to the advent of AlphaFold2, saw a progression towards more sophisticated modeling. Advances in computer hardware, including multi-core processors and GPUs, significantly revolutionized the landscape of computational biology and medicinal research, enabling more complex simulations and predictions . These advancements facilitated the development of methods for predicting native protein structures, simulating their dynamic behavior, and conducting Quantitative Structure-Activity Relationship (QSAR) studies, which are crucial for drug design . Computational biology and computer-aided drug design (CADD) specifically aid in characterizing ligand-binding mechanisms, identifying binding/active sites, and refining protein structures, thereby expediting the drug discovery process by reducing both time and financial costs and enhancing success rates in clinical trials .
The availability of accurate protein structures offers manifold benefits for drug design. Crucially, it enables the precise identification of potential drug binding pockets on the protein surface, facilitating the rational design of small molecules that can selectively interact with these sites . This structural insight allows for the design of specific inhibitors or activators, optimizing their fit, affinity, and selectivity, thereby minimizing off-target effects and potential toxicity . Furthermore, detailed structural information supports the understanding of molecular interactions at the atomic level, which is vital for lead optimization and the development of novel therapeutics with improved efficacy and safety profiles . By framing the state of the field before AlphaFold2, it becomes evident that while significant progress was made with experimental and early computational methods, the inherent limitations posed a continuous challenge to the efficiency and success of drug discovery.
1.2 AlphaFold2: A Paradigm Shift in Protein Structure Prediction and its Initial Impact on Drug Discovery
AlphaFold2 (AF2), developed by DeepMind and released in 2020, marks a transformative breakthrough in structural biology, effectively addressing the long-standing challenge of generating atomic-level protein structure models from sequence alone . This deep learning-based AI model, trained on extensive protein data, predicts three-dimensional protein structures with remarkable accuracy and speed, often comparable to experimental methods . AlphaFold2's accuracy in predicting human protein 3D structures is reported to be around 98.5%, a feat that has been lauded as the second-largest breakthrough in life sciences after the human genome project .
The "revolutionary impact" and "paradigm shift" claims for AlphaFold2 stem from its ability to overcome the significant limitations of previous methods for protein structure prediction . Traditionally, experimental techniques such as X-ray crystallography, Nuclear Magnetic Resonance (NMR) spectroscopy, and cryo-electron microscopy (cryo-EM) were time-consuming, laborious, and expensive, often taking years to determine a single protein structure . Early computational prediction methods like SWISS-MODEL, I-TASSER, and ROBETTA also faced limitations, including requirements for homologous structures or restrictions on sequence length, resulting in lower accuracy and reliability . In stark contrast, AlphaFold2 can provide rapid predictions, thereby democratizing access to structural data and enabling researchers to gain crucial insights into protein function and interactions at an unprecedented pace .
AlphaFold2's superior performance is primarily attributed to its deep learning methodology, particularly its utilization of novel attention-based neural network architectures. Unlike older methods that might predict contact maps, AlphaFold2 directly predicts atomic coordinates, a key advancement that contributed to its near-atomic precision in the 14th Protein Structure Prediction Competition (CASP14), where it significantly increased accuracy from 40 to 92.4 points . The model is trained on vast datasets from the Protein Data Bank (PDB) and UniProt, enabling accurate prediction of protein structures . The release of AlphaFold2's source code and a dataset of over 350,000 protein structures, including the human proteome, has further democratized access to predicted structures, making over 200 million protein structures accessible globally .
These capabilities initially impacted traditional drug discovery workflows by accelerating various stages of the process. By accurately predicting target protein structures, AlphaFold2 enables structure-based drug design (SBDD), allowing for the design of drugs that specifically target these proteins for more effective and safer treatments . This is particularly crucial for novel targets that lack experimentally determined structures, expanding the pool of accessible targets for drug development . For instance, AlphaFold has been successfully integrated into AI-driven drug discovery engines like Insilico Medicine's PandaOmics (for target selection) and Chemistry42 (for generative chemistry) to identify novel molecules for targets without experimental structures, such as a CDK20 small molecule inhibitor . The ability to predict protein-protein interactions is also valuable for understanding disease mechanisms and developing targeted therapies . This capability helps address bottlenecks in the traditionally inefficient drug discovery process, which often suffers from incomplete biological understanding and a lack of targets . However, it is important to note that while AlphaFold2 provides a static, highly accurate picture of the most stable protein conformation, it does not inherently capture the full range of protein dynamics or all possible shapes a protein can adopt, which is crucial for understanding drug interactions that involve conformational changes . Despite this, its accuracy, with pLDDT scores above 80 generally considered sufficient for SBDD purposes, significantly expedites the initial stages of drug design .
2. Advancements in Protein Structure Prediction Beyond AlphaFold2
The landscape of protein structure prediction has undergone a profound transformation, particularly with the advent of AlphaFold2, yet significant advancements are continuously emerging that extend capabilities beyond its initial scope .
This section provides a comprehensive overview of the sophisticated deep learning architectures and methodologies that have propelled the field forward, detailing their distinct principles and applications. It explores how these innovations address critical challenges, notably the limitations of predicting static protein structures by delving into the capture of protein dynamics and metastable conformations, which are paramount for understanding complex biological processes and rational drug design . Finally, the section delineates the rigorous benchmarking and performance evaluation strategies employed to assess the accuracy, speed, and utility of these advanced models, highlighting their implications for accelerating small molecule drug discovery . The integration of these cutting-edge methodologies signifies a paradigm shift from static predictions to a more dynamic, comprehensive understanding of protein function and interaction, crucial for future therapeutic development.
2.1 Deep Learning Architectures and Methodologies
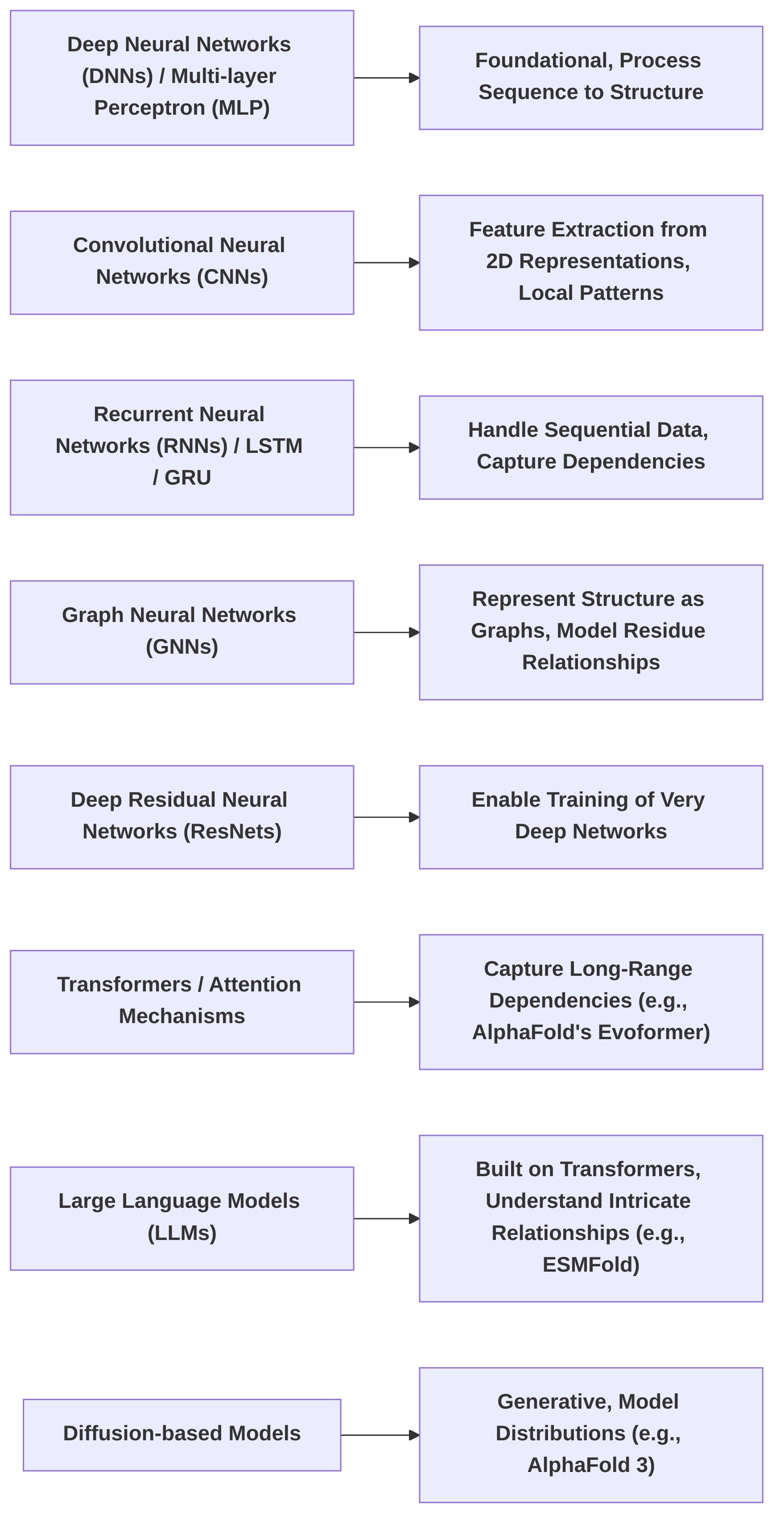
Recent advancements in protein structure prediction have been significantly driven by deep learning techniques, which analyze extensive datasets of known protein structures to predict unknown ones . Various deep learning architectures have been employed, each with distinct core principles and applications in this field .
Deep Neural Networks (DNNs) and Multi-layer Perceptron networks (MPLs) serve as foundational architectures, processing amino acid sequences to output structural predictions. While capable of learning complex distributions, their direct application to highly intricate protein structures can be limited due to their inherent feedforward nature .
Convolutional Neural Networks (CNNs) extract features from 2D representations of protein sequences using convolutional operations. These are particularly effective for local feature extraction, analogous to image processing, making them suitable for identifying patterns within short sequence segments that correspond to secondary structural elements .
Recurrent Neural Networks (RNNs), including Long Short-Term Memory (LSTM) and Gated Recurrent Units (GRUs), are designed to handle sequential data, processing protein sequences along their evolutionary or linear progression. Their ability to maintain a 'memory' of previous inputs makes them well-suited for capturing dependencies within the protein sequence, essential for understanding how distant residues interact .
Graph Neural Networks (GNNs) abstract protein structures as graphs, where amino acids are nodes and their interactions are edges. This representation allows GNNs to predict backbone geometry by modeling relationships between residues, making them powerful for capturing spatial relationships crucial for tertiary structure prediction .
Deep Residual Neural Networks (ResNets) address the challenge of training very deep networks by incorporating skip connections, enabling the training of models with hundreds or thousands of layers. This depth allows ResNets to learn increasingly complex hierarchical features from protein data .
Transformers, characterized by their attention mechanisms, have revolutionized protein structure prediction by excelling at capturing long-range dependencies within protein sequences, overcoming some limitations of RNNs. AlphaFold, for instance, employs an attention-based neural network architecture, specifically the Evoformer, a variation of the Transformer . Large Language Models (LLMs), built on the Transformer architecture, leverage vast parameter counts to understand intricate relationships in protein sequences, drawing parallels to natural language processing for sequence understanding . ESMFold, for example, is built on a BERT-like architecture (Transformer encoder layers) and uses masked residue prediction, enabling it to infer structural information from sequences without relying on Multiple Sequence Alignments (MSAs) or known homologies .
Diffusion-based models are generative models capable of modeling multimodal data distributions. These models are increasingly utilized for protein structure prediction and even novel protein design, demonstrating their ability to generate diverse and realistic protein conformations . AlphaFold 3, for instance, utilizes a diffusion network to assemble predictions, similar to those employed in AI image generation . This design represents a simpler architecture with fewer separate components compared to its predecessor, AlphaFold2 .
AlphaFold2, a landmark in protein structure prediction, significantly advanced the field through its innovative deep learning architecture, particularly the Evoformer module. While its specific architecture is complex, it is rooted in attention mechanisms, enabling it to capture intricate relationships within protein sequences and between homologous sequences. AlphaFold2's primary output includes highly accurate 3D protein structures, alongside per-residue confidence scores (pLDDT) and predicted alignment error (PAE) matrices. These outputs provide not only the predicted structure but also a robust measure of its accuracy and certainty, which is crucial for downstream applications like drug discovery. For instance, AF2BIND leverages AlphaFold2's "pair representation," a tensor assigned to each pair of amino acids predicting their relative positions, to infer ligand binding sites. This representation, which includes pairwise attention embeddings between target protein residues and appended "bait" amino acids, is fed into a logistic regression model. This approach demonstrates a key output type of AlphaFold2: latent representations that capture deep structural and interaction information, even beyond the direct 3D coordinates .
Beyond AlphaFold2, other notable hybrid deep learning methods include RoseTTAFold, which combines neural networks with energy-based approaches and has been updated to model complete biological assemblies . RoseTTAFold employs a "three-track" neural network that integrates sequential patterns, amino acid interplay, and probable 3D configurations. OpenFold, an open-source, trainable version of AlphaFold2, matches its prediction quality while offering faster processing and lower memory usage, highlighting efforts to democratize and optimize these powerful tools .
The transferability of learned features between different architectures and protein families is a critical aspect for the generalizability of these models. For example, the success of Transformer-based models like ESMFold, which learns structural information directly from sequences without MSAs, suggests that powerful, generalizable representations can be learned from vast unsupervised protein sequence datasets. Similarly, the ability of AF2BIND to leverage AlphaFold2's internal pair representation for a different task like binding site prediction indicates that features learned for structure prediction are highly transferable and contain rich information about protein interactions . This transferability allows for the development of new applications and fine-tuning of models for specific tasks (e.g., small proteins, large complexes, membrane proteins) without retraining from scratch, significantly accelerating research.
These advancements contribute to a more comprehensive understanding of protein structures beyond what AlphaFold2 alone provides. While AlphaFold2 excels at predicting single protein structures, the evolution of architectures like AlphaFold 3 with diffusion models and tools like RoseTTAFold, ESMFold, and OpenFold, alongside novel protein design tools like ProGen, ProteinMPNN, EvoDiff, and RFdiffusion, demonstrates a broader scope. These tools are increasingly capable of modeling complex biological assemblies, understanding protein-ligand interactions, and even designing novel proteins from scratch, moving beyond merely predicting static structures to enabling dynamic and functional insights crucial for small molecule drug discovery . This signifies a shift towards more versatile and integrative AI solutions that can inform various stages of the drug discovery pipeline.
2.2 Capturing Protein Dynamics and Metastable Conformations: Addressing Limitations of Static AlphaFold2 Predictions
While AlphaFold2 (AF2) has revolutionized static protein structure prediction, a significant limitation lies in its inability to comprehensively capture protein dynamics and predict metastable conformational states, which are crucial for understanding ligand binding and drug efficacy . AF2 typically predicts a single, often apo (unbound) conformational state, overlooking the conformational changes enzymes undergo upon ligand binding, the existence of cryptic binding sites, or the diversity arising from mutations and post-translational modifications . This static output is particularly insufficient for targets with multiple domains where ligand binding may occur at domain interfaces .
To address these limitations, several advanced approaches have emerged to incorporate protein dynamics into structure prediction. One such method is AlphaFold2-RAVE (AF2RAVE), which combines AF2 with all-atom enhanced sampling molecular dynamics and induced fit docking (Glide) . AF2RAVE specifically aims to overcome AF2's oversight of ligand-induced holo structures and critical metastable conformations. For instance, it can sample metastable states like the DFG-out conformation, which is essential for binding type II kinase inhibitors, a state not readily predicted by standard AF2 . This integrated AF2RAVE-Glide workflow has demonstrated considerable success, achieving over 50% success rates in docking known type II kinase inhibitors to metastable conformations of kinases such as Abl1, DDR1, and Src, thereby enhancing structure-based drug design (SBDD) for conformation-selective ligands .
Another innovative approach is the subsampled AlphaFold2 method, developed by Brown University researchers . This technique manipulates AF2 by feeding it smaller, subsampled portions of the protein sequence, rather than the entire sequence at once. This "confuses" the model, encouraging it to explore and predict a wider range of possible protein conformations . By rapidly predicting multiple protein configurations and their population frequencies, this method moves beyond static models to offer a more dynamic, "4D" understanding of proteins, including changes over time. This capability is crucial for identifying multiple ways to target proteins with drugs, as drug efficacy is highly dependent on the specific protein shape at the binding site, which can vary with conformational changes .
The difficulty in capturing protein dynamics with current AI models, including earlier versions of AlphaFold, stems from their primary training objective of predicting a single, most stable conformation from static sequence information . While AlphaFold2 itself is robust to small backbone changes (within ~1Å RMSD) and insensitive to sidechain dihedral angles for predicting binding sites, it does not inherently model large-scale conformational transitions . Such dynamic transitions, including ligand-induced fit effects and the exploration of metastable states, are complex and require moving beyond single-snapshot predictions.
Looking forward, models like AlphaFold 3 represent a significant step in expanding the scope of structure prediction. AlphaFold 3 can predict not only protein structures but also their interactions with a diverse array of biological entities, including DNA, RNA, ligands, ions, and various chemical modifications . This broader capability provides a more holistic understanding of biological processes and significantly enhances the accuracy of predicting protein-small molecule interactions, with reported accuracy increases of at least 50%, and even doubling for some critical interaction categories . This expanded predictive power is transformative for drug discovery, allowing for more precise identification of druggable pockets and optimization of therapeutic candidates based on complex molecular interactions . However, even AlphaFold 3, while revolutionary in its interaction predictions, still primarily focuses on static conformations and may not fully capture dynamic transitions or fold-switching proteins, exhibiting limitations such as chirality mismatches or steric clashes in some complex predictions . Therefore, continued efforts integrating dynamic sampling methods with advanced AI models will be essential for realizing the full potential of protein structure prediction in drug discovery.
2.3 Benchmarking and Performance Evaluation
The evaluation of protein structure prediction quality relies on a comprehensive set of metrics and rigorous benchmarking strategies. A primary benchmark is the Critical Assessment of Structure Prediction (CASP) competition, an annual event where researchers submit models for proteins with undisclosed experimental structures. The results are meticulously assessed and published, serving as a critical indicator of advancements in the field . Beyond CASP, various metrics are employed to quantify prediction accuracy. These include Q3 and Q8 accuracy for secondary structure prediction, Root Mean Square Deviation (RMSD) for global structural similarity, TM-score for topological similarity, and Global Distance Test Total Score (GDT_TS) for overall structural quality . For local structural assessment, LDDT/pLDDT scores are utilized, with pLDDT scores above 80 generally deemed necessary for reliable in silico modeling and virtual screening in drug discovery . DockQ Score specifically evaluates protein-protein docking accuracy . In the context of virtual screening, metrics like the enrichment factor are used to evaluate the effectiveness of computational methods, augmented by binding free energy calculations (e.g., MM-PBSA and MM-GBSA) and QM/MM scoring functions to enhance docking pose accuracy and binding affinity . Cross-validation techniques, such as hold-out, k-fold, and leave-one-out (LOOCV), are also fundamental for model validation .
AlphaFold2 marked a significant leap in protein structure prediction, achieving a GDT_TS score of 92.4 points in CASP14, substantially surpassing previous methods . Approximately two-thirds of AlphaFold2's predictions were comparable in accuracy to experimentally determined structures . This level of accuracy, often compared to crystallographic data at 2.0 Å resolution, is considered sufficient for structure-based drug design (SBDD) .
While AlphaFold2 set a high bar, subsequent models have emerged with distinct advantages and challenges. ESMFold, for instance, exhibits significantly faster prediction speeds (e.g., 6x faster than AlphaFold2) despite having fewer parameters. However, its mean GDT-TS score of 61.62 is lower than AlphaFold2's 73.06, indicating a trade-off between speed and accuracy. RoseTTAFold generally performs below ESMFold in over 80% of cases, whereas OpenFold matches AlphaFold2's prediction quality while being slightly faster .
Challenges persist, particularly in predicting protein dynamics and specific conformational states crucial for drug discovery. Standard AlphaFold2 structures often prove insufficient for docking type II inhibitors, which target specific DFG-out states, resulting in high ligand RMSDs. Even methods like rMSA AF2, designed to generate diverse structures, struggle to reliably sample these metastable states . To address this, AF2RAVE integrates enhanced sampling and a physics-based protocol (SPIB) to enrich holo-like structures from ensembles, significantly improving docking success rates, as evaluated by ligand RMSD . Similarly, while subsampled AlphaFold2 predictions have been validated against experimental data like NMR for specific proteins, the broader applicability and independent validation methods for these dynamic predictions remain an area for further development .
Newer models are also pushing boundaries in interaction prediction. AF2BIND, which leverages AlphaFold2's pair representation, demonstrates superior performance in binding-site prediction compared to other representations (AF2 single, ESM2, ESM1-IF), achieving a 66% binding-site residue recovery rate, which marginally increases to 69% when combined with ESM2 and ESM1-IF embeddings . This performance, akin to contact accuracy metrics, is rigorously evaluated on meticulously split datasets to prevent data leakage and ensures that predicted binding probabilities correlate with recovery . AF2BIND has shown accurate predictions on held-out protein classes and can even flag potentially spurious ligand placements in experimental structures that correlate with poor crystallographic data quality . The most recent advancement, AlphaFold 3, claims unprecedented accuracy in predicting molecular interactions, including protein-ligand and antibody-protein binding, surpassing all existing systems. On the PoseBusters benchmark, AlphaFold 3 is reported to be 50% more accurate than traditional methods, establishing itself as the first AI system to outperform physics-based tools for biomolecular structure prediction without requiring structural input .
These performance differences have significant implications for drug discovery. While AlphaFold2's high accuracy in static structure prediction has made it invaluable for initial target identification and virtual screening, the limitations in sampling dynamic conformations highlight the need for specialized tools like AF2RAVE for targets requiring specific conformational states. The emergence of AF2BIND and AlphaFold 3, with their enhanced capabilities in predicting ligand binding sites and molecular interactions, promises to revolutionize early-stage drug discovery by improving the accuracy of lead identification and optimization, potentially reducing the reliance on extensive experimental validation for binding poses. The continuous development and benchmarking of these models underscore the dynamic nature of the field and its profound impact on accelerating drug discovery workflows.
3. Impact on Small Molecule Drug Discovery Workflows

The advent of advanced protein structure prediction (PSP) models, particularly AlphaFold2 (AF2) and its subsequent iterations, has fundamentally reshaped small molecule drug discovery workflows by significantly accelerating and enhancing multiple critical stages, from target identification to lead optimization and high-throughput screening . By providing highly accurate three-dimensional protein structures and insights into protein dynamics, these computational tools are enabling researchers to bypass time-consuming experimental procedures, thereby streamlining the drug discovery pipeline and conserving resources .
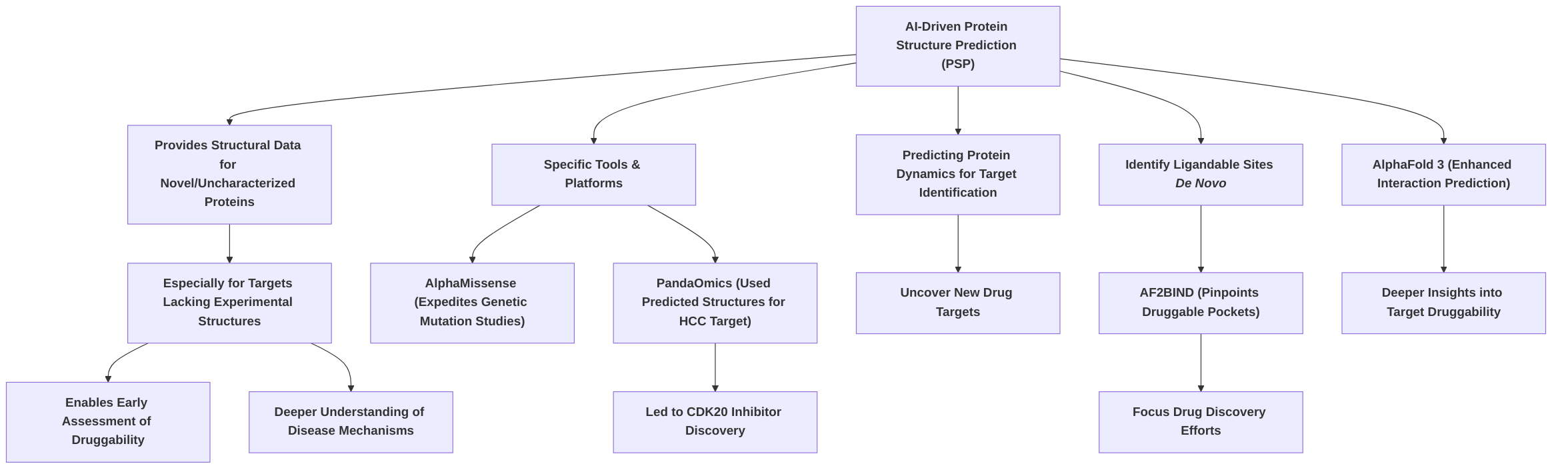
In the initial phase of Accelerated Target Identification and Validation, AI-driven PSP models provide crucial structural data for novel or previously uncharacterized proteins, especially those lacking experimental structures, thus enabling early assessment of druggability and a deeper understanding of disease mechanisms . Tools like AlphaMissense expedite genetic mutation studies for target identification, while platforms such as PandaOmics have successfully leveraged predicted structures to identify novel small molecule inhibitors, as demonstrated by the discovery of a CDK20 inhibitor for hepatocellular carcinoma . Furthermore, the ability to predict protein dynamics and identify ligandable sites de novo with tools like AF2BIND is critical for uncovering novel drug targets and focusing drug discovery efforts on promising therapeutic intervention points .
The impact extends significantly to Enhanced Structure-Based Drug Design (SBDD) and Lead Optimization. Accurate protein structures from AlphaFold2 facilitate the precise design of small molecules to modulate protein function, allowing for structure-based virtual screening (SBVS) of vast compound libraries . While standard AlphaFold2 predictions often represent static conformations, advanced workflows like AF2RAVE-Glide address this limitation by modeling metastable protein conformations crucial for designing conformation-selective ligands, significantly improving docking success rates for challenging targets like type II kinase inhibitors . This dynamic understanding, coupled with tools like AF2BIND for identifying binding residues, directly informs lead compound modifications to enhance binding affinity and specificity, thereby accelerating the optimization process .
In High-Throughput Virtual Screening and Hit Identification, the synergy between AlphaFold2's rapid, accurate protein structure predictions and large chemical libraries like ZINC20 has revolutionized the process. This integration enables the screening of billions of compounds in unprecedented timelines, dramatically increasing hit rates and reducing reliance on experimental methods . AlphaFold2-generated models provide essential structural templates for SBVS, while confidence scores (pLDDT) from these models can guide screening efforts. Further advancements, such as AF2RAVE's ability to enrich holo-like structures, have substantially increased docking success rates, making large-scale virtual screening campaigns more productive. Real-world applications, such as the rapid identification of a novel CDK20 hit compound, underscore the efficiency gains .
While PSP models provide the structural foundation, AI for De Novo Drug Design and ADMET Prediction complements these efforts by generating novel small molecules with desired properties. Generative chemistry platforms, such as Chemistry42, leverage predicted protein structures to create new compounds tailored to specific targets, moving beyond the optimization of existing molecules . AI models employ various representations and deep learning techniques to optimize properties like binding affinity and drug-likeness (QED), contributing to a more efficient and cost-effective drug development pipeline. Though the direct link between PSP and ADMET prediction is sometimes indirect, accurate protein structures can inform molecule design to minimize off-target interactions and anticipate ADMET issues early in development, with the ultimate goal of generating new chemical entities that possess favorable ADMET characteristics and synthetic accessibility .
Illustrative Examples of AI-Accelerated Drug Discovery further solidify the transformative impact, showcasing tangible outcomes beyond theoretical promise. The identification of the CDK20 inhibitor ISM042-2-048 marks a direct application of AlphaFold in hit identification, while platforms like Insilico Medicine's Pharma.AI have led to significant milestones like FDA Orphan Drug Designation . The AF2RAVE-Glide workflow's success in accurately modeling and targeting challenging metastable conformations of kinases, achieving over 50% success rates for docking type II inhibitors, quantifies the acceleration in conformation-selective drug discovery . Furthermore, computational optimizations to AlphaFold2, resulting in a 22-fold speedup, significantly reduce the computational burden, enhancing the overall efficiency of the drug discovery pipeline .
Despite these profound advancements, challenges persist across all these workflows. The accurate prediction of intrinsically disordered proteins and highly flexible regions, capturing full protein dynamics, and seamlessly integrating predicted structures into existing complex computational workflows (e.g., molecular dynamics, QM/MM) remain areas requiring continuous improvement and refinement . Similarly, while AI excels at de novo design, the reliability and generalizability of ADMET predictions still face limitations due to the inherent complexity of biological systems. Future directions will focus on addressing these challenges through enhanced validation strategies, improved dynamic modeling, and the development of more robust, integrated AI solutions capable of navigating the intricate landscape of drug-target interactions and whole-system biological responses.
3.1 Accelerated Target Identification and Validation
The advent of AlphaFold2 (AF2) and other advanced AI models has profoundly accelerated the process of target identification and validation, particularly for proteins where experimental structural data remain elusive . By providing highly accurate predictions of protein structures, these computational tools enable a deeper understanding of disease mechanisms and facilitate the discovery of novel drug targets . This acceleration is critical for drug discovery pipelines, as it allows researchers to bypass the often time-consuming and resource-intensive experimental determination of protein structures .
One significant benefit of AF2 and similar AI models is their ability to identify and validate novel drug targets, especially those previously poorly understood or lacking substantial structural information . For instance, AF2 predictions offer three-dimensional data for newly identified targets, particularly in pathogens where experimental structures are unavailable, thereby enabling early assessment of druggability. Key factors for prioritizing these targets include the confidence level (pLDDT score) of the prediction, the size and accessibility of predicted binding pockets, and the uniqueness of the protein fold . Furthermore, these predicted structures are instrumental in understanding protein function, identifying potential druggable pockets, and validating targets through structural analysis .
Specific examples illustrate the practical benefits of this acceleration. AlphaMissense, an AI tool leveraging AF2 structural insights, can efficiently select genetic mutations for study, thereby expediting novel drug target identification . In a concrete demonstration of target validation, the biocomputational platform PandaOmics utilized AI-powered predicted structures to identify a protein of interest for hepatocellular carcinoma (HCC) treatment, ultimately leading to the efficient discovery of a novel CDK20 small molecule inhibitor . This exemplifies how AI-driven predictions can lead to successful target identification and validation, even when experimental data is scarce.
Beyond static structures, the capacity to predict protein dynamics and multiple conformations represents a significant advancement for target identification. Understanding the conformational landscape of a protein allows scientists to design drugs that bind more effectively to active protein configurations, leading to more potent and specific therapeutic effects . This dynamic insight can uncover new drug targets that might have been overlooked due to their static conformations not appearing as viable candidates, especially critical in areas like targeted cancer therapy where precise targeting of specific protein configurations is essential .
The capability to identify ligandable sites de novo is another crucial aspect of accelerated target identification. Tools like AF2BIND, which accurately predict small-molecule-binding residues on newly predicted or uncharacterized protein structures, can highlight potential therapeutic intervention points. The synergy between AF2-predicted structures and AF2BIND's ability to pinpoint druggable pockets allows researchers to focus drug discovery efforts on promising targets without requiring known homologous structures or experimental data . The recent development of AlphaFold 3, with its enhanced ability to predict interactions between proteins and a wide spectrum of biomolecules, including ligands, further amplifies this capability by providing deeper insights into target druggability and accelerating the design of next-generation treatments .
While these advancements offer immense potential, challenges remain. The accuracy of predictions, especially for intrinsically disordered proteins or highly flexible regions, requires continuous improvement. Additionally, integrating predicted structures seamlessly into existing drug discovery workflows and ensuring their reliable use in downstream applications, such as virtual screening and lead optimization, necessitates further development. These challenges and potential solutions, including refinement techniques and enhanced validation strategies, will be elaborated upon in subsequent chapters. Moreover, predicted structures can accelerate the process of obtaining refined experimental structures by providing reliable starting models for fitting into cryo-EM or X-ray data, thereby bridging the gap between computational and experimental approaches . Computational biology approaches, encompassing structural biology, molecular biology, genomics, and bioinformatics, alongside techniques like molecular mechanics (MM), quantum mechanics (QM), and molecular dynamics (MD) simulations, are being widely explored to understand pathogenesis and identify targets, reinforcing the foundational role of accurate protein structures, whether predicted or experimental, in drug discovery .
3.2 Enhanced Structure-Based Drug Design (SBDD) and Lead Optimization
AlphaFold2's remarkable accuracy in protein structure prediction significantly enhances the efficiency and success rate of Structure-Based Drug Design (SBDD) techniques. By providing precise three-dimensional blueprints of target proteins, AlphaFold2 accelerates the identification of small molecules or antibodies capable of binding to specific proteins and modulating their function, thereby expediting drug discovery and development . Researchers can leverage these predicted structures to design drugs that precisely fit into target protein active sites, either enhancing or inhibiting their activity . This predictive power enables structure-based virtual screening (SBVS) of extensive compound libraries to pinpoint potential drug candidates, as the availability of accurate target structures facilitates the identification of binding pockets and functional regions . For instance, AlphaFold has been successfully integrated with generative chemistry platforms like Chemistry42 to identify novel hit molecules, as exemplified by the discovery of a potent CDK20 inhibitor, ISM042-2-048, with a Kd value of 566.7 ± 256.2 nM and an IC50 of 33.4 ± 22.6 nM, showcasing a substantially accelerated SBDD workflow . AlphaFold 3 further extends these capabilities by predicting interactions involving drug-like molecules, including ligands and antibodies, with unprecedented accuracy, directly contributing to drug design efforts .
While AlphaFold2 excels at predicting static protein structures, its primary limitation in SBDD is its inability to inherently model protein dynamics and diverse conformational states that are crucial for ligand binding and selectivity. Traditional AlphaFold2 predictions often represent a single, stable conformation, which may not be the most relevant state for ligand interaction. However, advanced models and workflows built upon AlphaFold2 address this. For example, the AF2RAVE-Glide workflow specifically enhances SBDD by enabling the modeling of metastable protein conformations, which are critical for designing conformation-selective ligands . This approach allows for the exploration of alternative protein states that can be stabilized upon ligand binding, directly addressing the static nature of standard AlphaFold2 predictions. For instance, AF2RAVE-Glide has successfully docked type II kinase inhibitors to DFG-out conformations of protein kinases, demonstrating its utility in rational drug design and optimization, particularly for targets requiring selectivity through conformation targeting . This ability to generate holo-like structures with sufficient accuracy for subsequent docking represents a significant leap from relying solely on static, experimentally determined structures or less reliable free energy perturbation methods for small molecules without exemplars .
The deeper understanding of protein dynamics and interactions derived from advanced models like AF2RAVE allows for more intelligent lead optimization. Dynamic protein insights directly inform lead compound modifications to enhance binding affinity and specificity. The ability to predict the conformational landscape of proteins is a "game-changer" for drug discovery, enabling the design of drugs that bind more readily to active conformations, leading to more potent and specific therapeutic effects . This dynamic understanding can also unveil new drug targets previously overlooked due to their static conformations not appearing as favorable. Unlike merely stating that dynamic information aids optimization, models like AF2RAVE directly inform design principles by providing conformation-selective insights. Specifically, AF2RAVE addresses the challenge of designing conformation-selective ligands by generating and docking to metastable protein conformations . While specific quantifiable improvements in docking success rates or RMSD achieved by AF2RAVE compared to standard AlphaFold2 were not explicitly detailed as numerical values in the provided digests, the successful docking of type II kinase inhibitors to DFG-out conformations, a challenging task for static models, implicitly underscores its enhanced accuracy and specificity . This demonstrates a tangible acceleration of the drug discovery process by reducing computational time and allowing for a more focused approach in hit identification and lead optimization .
Further aiding lead optimization, models such as AF2BIND directly contribute to SBDD by providing residue-level probabilities of ligand interaction. This allows for a more focused approach to identifying and characterizing binding pockets, guiding the optimization of lead compounds by suggesting key interaction points . The analysis of "bait" residue contributions, which correlate with ligand hydrophobicity, offers insights into the chemical nature of the binding site, potentially aiding in the design of complementary small molecules . AF2BIND's output provides a hierarchy of residues most likely involved in binding, offering more actionable information than purely volumetric pocket-finding algorithms .
Despite these advancements, challenges remain. AlphaFold2 predictions, by themselves, do not include ligands, necessitating further work for hit identification and lead optimization . While methods like Free Energy Perturbation (FEP) can estimate binding affinity, their reliability for small molecules without exemplars is noted as lower . The integration of dynamic protein insights into existing computational workflows, such as molecular dynamics (MD) simulations and quantum mechanics/molecular mechanics (QM/MM) methods, which are crucial for capturing conformational changes and detailed ligand-target interactions, requires continuous refinement . These challenges and potential solutions will be elaborated upon in Chapters 4 and 5.
3.3 High-Throughput Virtual Screening and Hit Identification
The advent of advanced protein structure prediction (PSP) technologies, particularly AlphaFold2, in synergy with the exponential growth of vast chemical libraries, has initiated a transformative era in high-throughput virtual screening (HTVS) for drug discovery . This integration significantly enhances the identification of promising drug candidates, thereby reducing the reliance on costly and time-intensive experimental screening methods .
The core of this revolution lies in AlphaFold2's capability to rapidly predict protein structures with high accuracy at low cost, complemented by the immense scale of compound databases such as ZINC20 . ZINC20 alone boasts nearly 2 billion compounds, with 1.3 billion readily available for purchase, and the potential for tens of billions of custom-built compounds exhibiting substantial scaffold and molecular diversity . This unprecedented scale, coupled with efficient computational platforms like VirtualFlow, which can screen 1 billion compounds in 15 hours using 160,000 CPUs, dramatically increases hit rates, accelerates computations, and strengthens iterative screening capabilities, thereby overcoming the previously daunting timeline of 475 years for screening 1 billion compounds on a single CPU . Companies like MCE further leverage this synergy through professional virtual screening services, offering access to over 40 high-throughput compound libraries comprising approximately 6 million purchasable, reproducible, and diverse drug-like compounds .
The efficacy of virtual screening is critically dependent on the accuracy of the protein structures employed. AlphaFold2-generated models provide accurate structural templates, which are pivotal for structure-based virtual screening (SBVS) by facilitating the identification of potential binding pockets and functional regions on target proteins . This enables the in silico exploration of candidates that bind to specific targets, thereby improving the efficiency and reducing the expenditure of the hit-to-lead process . While many traditional PSP papers do not explicitly detail their use in HTVS , the general consensus supports that accurate protein modeling combined with protein-ligand docking is highly beneficial for virtual screening . Tools like AF2BIND, by accurately predicting small-molecule binding sites, even on proteins without prior structural data, directly support HTVS by prioritizing targets and refining docking strategies . AF2BIND's ability to provide a hierarchy of likely binding residues can further inform scoring functions and focus virtual screening efforts on specific regions, making large-scale identification of binding sites across vast proteomes feasible .
Significant improvements have been observed when integrating refined predicted structures into virtual screening workflows. For instance, the use of AF2RAVE to enrich holo-like structures significantly enhanced the success rate of docking known type II inhibitors from very low with unrefined AlphaFold2 structures to over 50%, making large-scale virtual screening campaigns more productive . This contrasts with instances where AlphaFold2 predictions alone may not capture dynamic conformational changes critical for ligand binding, highlighting the importance of methods that refine these structures or consider ensembles of conformations.
The predictive uncertainty or confidence scores inherent in advanced PSP models can be strategically integrated into virtual screening workflows to prioritize hits and filter out lower-confidence predictions. For example, AlphaFold2 provides per-residue confidence scores (pLDDT) which can indicate the reliability of a predicted structure, guiding decisions on whether to proceed with virtual screening on a particular target. Although specific integration methodologies for these confidence scores into virtual screening workflows are not extensively detailed in all digests, the general principle suggests that higher confidence scores would lead to more reliable docking results. For instance, AlphaFold 3's high accuracy in predicting ligand-protein interactions, surpassing existing methods, intrinsically supports enhanced virtual screening by improving the quality of the structural input .
The efficiency gains are evident in real-world applications; for example, a novel CDK20 hit compound was identified within 30 days from target selection with only seven compounds synthesized, a feat attributed to AlphaFold predictions combined with generative chemistry tools . This process further yielded a more potent hit after an initial round of AI-powered compound generation and testing . Moreover, optimizations in AlphaFold2 processing times, achieving 22x faster benchmarks on the same GPU hardware, implicitly support the acceleration of virtual screening by speeding up tasks related to hit identification .
Despite these advancements, challenges remain. The dynamic nature of protein conformations, which are crucial for ligand binding, is not always fully captured by static predicted structures. This necessitates the integration of techniques like Molecular Dynamics (MD) simulations to improve conformational sampling and QM/MM approaches to enhance scoring in virtual screening workflows . Furthermore, while deep learning methods such as EquiBind, GNINA, and DiffDock are significantly changing the virtual screening landscape with their speed and accuracy in predicting binding poses , continuous refinement is needed to address limitations in predicting flexible binding sites or accurately modeling induced fit mechanisms. These challenges and their potential solutions will be elaborated upon in subsequent chapters, specifically Chapter 4 and Chapter 5.
3.4 AI for De Novo Drug Design and ADMET Prediction
AI-driven compound generation plays a crucial role in complementing protein structure prediction by enabling the design of novel small molecules with desired properties. While many recent advancements in protein structure prediction, such as AlphaFold 2 and AlphaFold 3, primarily focus on predicting the structures and interactions of existing molecules, including ligands, they do not directly detail de novo drug design or ADMET prediction . However, the availability of highly accurate protein structures, often derived from these prediction methods, provides a fundamental basis for AI algorithms to design novel small molecules.
One significant success in this interplay is exemplified by the use of generative chemistry platforms, such as Chemistry42, which can leverage predicted protein structures to generate novel molecular entities through de novo design . This approach allows for the creation of new compounds tailored to specific protein targets, moving beyond the optimization of existing molecules. Although a direct, detailed application of ADMET prediction within this specific context is not always explicitly elaborated in some literature, the overarching goal of de novo design implicitly involves optimizing various molecular properties, including those related to ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) .
The broader AI-driven optimization landscape extensively utilizes generative models to create novel molecules with desired characteristics. These models often employ various representations, including SMILES-based, graph-based, and 3D-based, alongside deep learning techniques such as Variational Autoencoders (VAEs), Generative Adversarial Networks (GANs), and Normalizing Flows . These AI models are trained to learn molecular distributions and can be guided, frequently through reinforcement learning, to optimize properties like binding affinity, quantitative estimate of drug-likeness (QED), and lipophilicity (logP), which are crucial for drug efficacy and safety. The ability of AI to optimize molecular properties in input, latent, and output spaces directly contributes to a more efficient and less costly drug development pipeline .
While the focus on direct connections between protein structure prediction and ADMET property prediction is sometimes indirect in the provided digests, the general trend of AI integration in drug design strongly implies its importance. Accurate protein structures can inform the design of molecules that better fit target binding sites, which in turn can influence their ADMET profiles. For example, a molecule designed to tightly bind a specific target might inadvertently interact with off-targets, leading to toxicity. AI-driven ADMET prediction models, when combined with accurate protein structures, can help anticipate such interactions, contributing to a more holistic and efficient drug discovery process by identifying potential issues early in the development pipeline. The goal is to generate new chemical entities that not only exhibit high binding affinity but also possess favorable ADMET characteristics and synthetic accessibility .
Despite the promising advancements, current AI models for predicting complex biological interactions, including ADMET properties, still face limitations in terms of reliability and generalizability. Predicting the intricate interplay between a drug candidate and the vast biological system, including absorption rates, distribution patterns, metabolic pathways, and excretion routes, is challenging due to the inherent complexity and variability of biological systems. The reliability of these predictions can impact drug safety and efficacy, potentially leading to costly failures in later stages of development. While generative AI tools for protein sequences and structures, such as ProGen, ProteinMPNN, EvoDiff, and RFdiffusion, show promise for generating novel therapeutic proteins , the application of AI for de novo small molecule design and ADMET prediction is a distinct, though related, field that requires specialized literature for a comprehensive analysis of its specific challenges and successes. The continued development of robust AI models, coupled with experimental validation, is essential to overcome these limitations and fully realize the potential of AI in accelerating drug discovery.
3.5 Illustrative Examples of AI-Accelerated Drug Discovery
The advent of AI, particularly advanced protein structure prediction models like AlphaFold2, has demonstrably accelerated various stages of small molecule drug discovery, transitioning from theoretical promise to tangible outcomes . One significant acceleration lies in the streamlining of protein structure prediction itself, allowing researchers to rapidly acquire target structures and thereby focus on developing promising drug candidates, which also helps optimize drug design by predicting interactions with target proteins, ultimately conserving time and resources .
Concrete examples illustrate this acceleration. In a notable instance, AlphaFold was employed to predict the structure of CDK20, facilitating the identification of a novel small molecule inhibitor, ISM042-2-048, which exhibited high inhibitory activity (IC50 of 33.4 ± 22.6 nM) and selective anti-proliferative activity in HCC cells (IC50 of 208.7 ± 3.3 nM) . This marks the first documented application of AlphaFold in the hit identification phase, showcasing a direct impact on early-stage drug discovery. Beyond hit identification, AI platforms have demonstrated broader utility; for example, Insilico Medicine's Pharma.AI platform, which leverages AI for drug discovery and development, led to the FDA Orphan Drug Designation for INS018_055 . Similarly, AI has been instrumental in discovering new antibiotics like "halicin" and developing therapeutics for conditions such as obsessive-compulsive disorder (DSP-1181), and it played a crucial role during the COVID-19 pandemic by combining with fragment-based drug design to identify potential SARS-CoV-2 inhibitors . AlphaFold3, the latest iteration, further enhances predictive capabilities by modeling interactions of all life's molecules, as demonstrated by its accuracy in predicting viral spike protein interactions with antibodies and sugars, and protein-DNA complexes .
A critical bottleneck in drug discovery, particularly for kinase inhibitors, is the accurate modeling of metastable protein conformations. Standard AlphaFold2 structures often prove inadequate for docking type II kinase inhibitors, which target the DFG-out state . This limitation was specifically addressed by the AF2RAVE-Glide workflow. This workflow successfully modeled these critical metastable conformations, enabling the targeting of the DFG-out state for kinases such as Abl1, DDR1, and Src . For DDR1, the AF2RAVE workflow identified a "holo-model" from its rMSA AlphaFold2 decoys, which was critical for the successful docking of type II inhibitors with a low Root Mean Square Deviation (RMSD). For more challenging kinases like Abl1 and Src, characterized by conserved Multiple Sequence Alignments (MSAs), an AF2-template homology modeling approach, guided by the latent space derived from DDR1, was employed to generate relevant metastable conformations . This targeted approach yielded over 50% success rates in docking known type II inhibitors to these kinases, showcasing a substantial improvement over standard AlphaFold2, which typically fails to predict these specific metastable states, thus quantifying the "acceleration" achieved in conformation-selective drug discovery .
Beyond improved conformational sampling, computational efficiency has also seen significant acceleration. Optimizations to AlphaFold2, such as those implemented in collaboration with the Ellison Medical Institute (EMI), have resulted in a remarkable 22-fold speedup in processing times on identical GPU hardware, while maintaining full prediction accuracy . This rapid optimization, achieved within weeks, signifies a critical acceleration in the drug discovery pipeline by reducing the computational burden associated with protein structure prediction. Similarly, AF2BIND has demonstrated accurate prediction of binding residues for various protein classes, including G-protein coupled receptors (GPCRs) and bromodomains, and has provided mechanistic insights into ligand binding based on amino acid activations, even for proteins not seen during training, enhancing the generalizability and utility of AlphaFold-derived models .
Despite these advancements, challenges persist. While AlphaFold2 provides high confidence models for many proteins, like the SARS-CoV-2 cysteine protease 3CL M pro, comparison with ligand-bound forms remains crucial to fully understand conformational changes critical for drug binding . Furthermore, issues such as low success rates in predicting accurate antigen-antibody complexes for antibody discovery illustrate ongoing limitations . Understanding protein dynamics and conformational changes due to mutations also presents a challenge, though methods like subsampled AlphaFold2 are showing promise in this area . These challenges, particularly the need for accurate prediction of dynamic protein behavior and protein-ligand interactions, will be further explored in Chapters 4 and 5, where potential solutions and future directions will be discussed.
4. Challenges and Limitations in AI-Driven Drug Discovery
The advent of AI, particularly models like AlphaFold2 and its successors, has significantly advanced protein structure prediction, yet their application in small molecule drug discovery encounters notable challenges and limitations . This section delineates the core limitations stemming from the static nature of predicted structures and the models' inability to fully capture protein dynamics, alongside the critical issues of data requirements, model generalizability, and interpretability in drug discovery contexts.
A primary constraint lies in the propensity of advanced models like AlphaFold2 to predict a single, static protein conformation, which fundamentally overlooks the essential dynamic nature of proteins and their conformational plasticity vital for biological function and ligand binding . This static representation is insufficient for accurately capturing ligand-induced conformational changes, domain movements in multi-domain proteins, or identifying cryptic binding sites . Specifically, targeting metastable protein conformations, such as the DFG-out state in kinases crucial for type II inhibitors, remains a significant hurdle. AlphaFold2 ensembles and refined methods often fail to reliably sample these specific states, leading to low success rates and high ligand RMSDs in docking studies . Furthermore, these models face difficulties with intrinsically disordered proteins, membrane proteins, and complex protein assemblies . AlphaFold3, while an advancement, still contends with dynamic systems like fold-switching proteins and may introduce structural inaccuracies such as chirality mismatches or steric clashes, particularly in larger complexes . A key technical challenge is the precise quantification of binding affinities, which current AlphaFold2 models struggle to achieve, despite efforts by tools like AF2BIND to predict de novo binding sites . These limitations are rooted in the training data, primarily the Protein Data Bank (PDB), which largely comprises static, thermodynamically stable conformations, thus biasing models and potentially leading to overfitting . The computational expense of these models further restricts their broad application . Beyond technical aspects, intellectual property and patentability issues present non-technical challenges, requiring demonstrable human guidance in AI-assisted drug design to secure patent protection . Addressing these limitations necessitates innovative solutions, including integrating AI with classical simulation methods, incorporating reinforcement learning, and expanding PDB data to include more diverse conformational states .
Further challenges arise from the stringent data requirements for AI models, their generalizability to novel targets, and the interpretability of their predictions. The efficacy of AI models is highly dependent on high-quality and diverse training data, often sourced from extensive datasets like the PDB . However, the structural diversity within these databases is often insufficient to capture dynamic protein conformations, requiring expanded datasets that include transient states . The meticulous curation of datasets, exemplified by AF2BIND's reliance on highly filtered protein-ligand complexes, underscores the rigorous data preparation needed . Data limitations and biases directly impair the generalizability and robustness of AI predictions, particularly when applied to novel or less-studied targets. Rigorous data splitting is crucial to prevent overfitting and ensure generalizability to unseen folds . While protocols like AF2RAVE demonstrate promising generalizability by transferring latent spaces and sampling strategies across different kinases, challenges persist, such as limitations in the quantitative reliability of absolute free energy values () from umbrella sampling due to sampling inefficiencies . Finally, interpretability remains a critical concern, as many deep learning models function as "black-box" systems, obscuring their internal decision-making processes . This lack of transparency impedes trust and adoption, as researchers cannot easily validate predictions or fine-tune processes . While some models like SPIB and AF2BIND offer positive interpretability by aligning learned reaction coordinates with physical features or allowing analysis of individual amino acid contributions, respectively, the need for enhanced transparency in de novo drug design and for complex biological processes is paramount . Addressing these intertwined challenges in data requirements, generalizability, and interpretability is crucial for building confidence in AI-driven drug discovery and accelerating therapeutic development.
4.1 Limitations of AlphaFold2 and Advanced Models in Drug Discovery Contexts
While AlphaFold2 has revolutionized protein structure prediction, providing highly accurate static models, its direct applicability to small molecule drug discovery is subject to several crucial limitations . Unlike the robust quality of structures derived from homology modeling for drug design applications , AlphaFold2 primarily predicts a single, static protein conformation . This static representation fundamentally overlooks the dynamic nature of proteins, which often exhibit conformational plasticity essential for their biological function and ligand binding . Consequently, AlphaFold2 models are insufficient for capturing crucial ligand-induced conformational changes, accurately predicting domain movements in multi-domain proteins, or identifying cryptic binding sites .
Specifically, standard AlphaFold2 predictions have proven insufficient for targeting metastable protein conformations, such as the DFG-out state in kinases, which is critical for type II inhibitors. Ensembles generated by AlphaFold2, or refined methods like rMSA AF2 and AF2-cluster, frequently fail to reliably sample these specific metastable states, even exhibiting biases towards template structures. This limitation significantly impacts drug design, evidenced by the very low success rates and high ligand RMSDs observed when docking type II inhibitors to AlphaFold2-derived structures . Furthermore, AlphaFold2 struggles with intrinsically disordered proteins, membrane proteins, and complex protein assemblies . AlphaFold3, while an advancement, still contends with dynamic systems like fold-switching proteins and may produce structures with chirality mismatches or steric clashes, particularly in larger complexes . It also requires a "relatively good understanding of biology" for effective use, implying a knowledge gap for broad accessibility .
A key technical challenge is the accurate prediction of binding affinities, which AlphaFold2 models struggle to quantify precisely, a critical metric for drug discovery . While tools like AF2BIND attempt to bridge the gap by predicting de novo binding sites, they acknowledge limitations such as a theoretical recovery ceiling below 100% due to the absence of the true ligand in prediction, potential signal dilution for larger proteins, and current training limited to single-chain proteins up to 500 amino acids . These models also often show insensitivity to sidechain rotamers, which, while beneficial for ambiguous structures, omits crucial detail for dynamics or affinity .
The underlying causes of these limitations are multifaceted. The difficulty in predicting dynamic proteins stems from AlphaFold2's training data, primarily derived from the Protein Data Bank (PDB), which predominantly contains static, thermodynamically stable conformations, thereby limiting the representation of dynamic or multi-state proteins . This structural bias can lead to overfitting and memorization effects, resulting in modest success rates for known fold-switching cases and poor performance on novel conformations . The computational expense of AlphaFold2 and related advanced models also poses a significant barrier to accessibility and broader application . Even computationally intensive methods like Molecular Dynamics (MD) simulations, while improving docking accuracy, face challenges with large-scale or long-timescale exploration and may not fully address cases involving valence electron transfer, for which Quantum Mechanics (QM) calculations, though more expensive, are required .
Beyond technical hurdles, non-technical challenges, notably intellectual property (IP) and patentability, are critical considerations. Drugs developed with significant AI involvement may face eligibility issues for patent protection if human contribution is deemed insufficient . Biotech companies must demonstrate clear human guidance in AI-assisted design or develop proprietary AI systems to ensure patentability .
Addressing these limitations requires innovative solutions. To tackle dynamic protein prediction, approaches like "subsampled AlphaFold2" are being investigated to predict a wider range of conformations, though their generalizability still needs further research . Integrating AI models with classical simulation methods, such as enhanced sampling MD simulations, could provide a more comprehensive view of protein dynamics and binding events. Furthermore, drawing parallels from other AI domains, such as incorporating reinforcement learning for exploring conformational landscapes or generative adversarial networks (GANs) for novel binding site generation, could offer avenues for improvement, though GANs also face challenges such as continuous drift of learning parameters and potential output distortion . From a data perspective, expanding the PDB to include more diverse conformational states, particularly for dynamic and multi-state proteins, would be crucial for training more robust AI models . For non-technical challenges, developing clear guidelines and legal frameworks for AI-assisted drug discovery and fostering collaborations that ensure transparent documentation of human intervention in AI workflows will be paramount.
4.2 Data Requirements, Model Generalizability, and Interpretability
The efficacy of AI models in drug discovery, particularly in protein structure prediction, is inherently dependent on the availability of high-quality and diverse training data. A significant challenge lies in obtaining sufficient high-quality data, as AI models are often trained on extensive datasets such as the Protein Data Bank (PDB) . However, the accuracy of these tools is constrained by the structural diversity within these databases, necessitating expanded datasets that include transient states to better capture dynamic protein conformations . For instance, AF2BIND's development relied on a meticulously curated dataset of protein-ligand complexes from the PDB, involving stringent filtering criteria such as resolution and R-factor, highlighting the rigorous data preparation required . Similarly, the accuracy of subsampled AlphaFold2 predictions is influenced by the quality and completeness of protein sequence data .
Data limitations and potential biases directly impact the generalizability and robustness of AI predictions, particularly when models are applied to novel or less-studied targets. The importance of rigorous data splitting, based on sequence, structure, and pocket similarity, is crucial for ensuring model generalizability to unseen folds and preventing overfitting . While AlphaFold 3's capabilities are attributed to its "next-generation architecture and training that now covers all of life’s molecules" , specific details regarding its data requirements or generalizability improvements are not explicitly provided. Conversely, the AF2RAVE protocol demonstrates promising generalizability, as its latent space and sampling strategies, initially developed for DDR1, could be successfully transferred to Abl1 and Src kinases, significantly reducing the need for complete model retraining . This indicates a correlation between diverse and well-structured training datasets and improved performance on out-of-distribution targets. However, challenges persist, such as the limitations in quantitative reliability of absolute free energy values () from umbrella sampling due to sampling inefficiencies . ESMFold's ability to infer structural information from sequences without relying on multiple sequence alignments (MSAs) or known homologies suggests a potential for enhanced generalizability in contexts where such data is scarce .
Interpretability is a critical consideration for AI models in drug discovery, especially when making high-stakes decisions based on AI predictions. Many deep learning models are often characterized as "black-box" systems, making their internal decision-making processes opaque to human scrutiny . This lack of transparency can hinder trust and adoption, as researchers cannot easily trace predictions, validate steps, or fine-tune processes . The interpretability of models directly influences decision-making in critical drug discovery stages, as an inability to understand the rationale behind a prediction can lead to unaddressed biases or missed insights. For instance, the SPIB model's positive interpretability is noted because its learned reaction coordinates align with physical features like A-loop position and DFG-type . In contrast, the interpretability of AF2BIND is highlighted as a key advantage due to its logistic regression nature, allowing for the analysis of individual amino acid contributions to predictions, a stark contrast to the opaque nature of more complex AI models . Research into the interpretability of deep learning models is crucial for enhancing the understanding of complex biological processes like sequence-to-structure mapping . Furthermore, for de novo drug design, challenges such as ensuring synthesizability and the poor interpretability of normalizing flows underscore the need for improved transparency . Strategies aimed at improving transparency and trustworthiness include the integration of AI predictions with experimental techniques to provide a more comprehensive understanding of protein structures and functions . Ultimately, addressing data requirements, enhancing generalizability, and improving interpretability are paramount for bolstering confidence in AI-driven drug discovery decisions and accelerating the development of novel therapeutics.
5. Future Directions and Emerging Trends
The landscape of small molecule drug discovery is continuously evolving, driven by the rapid advancements in AI-driven protein structure prediction. The future trajectory of this field is characterized by the strategic integration of diverse data types, continued refinement and synergistic application of computational and experimental methodologies, and the proactive establishment of robust ethical and regulatory frameworks. This section outlines key emerging trends and future directions that will shape the next generation of drug discovery. It emphasizes the critical need for a holistic approach that transcends isolated computational predictions, embracing a more comprehensive understanding of biological systems and responsible technological deployment.
One primary future direction involves the Integration of Multi-Omics Data with Structural Biology. The proliferation of multi-omics data, encompassing genomics, transcriptomics, proteomics, and metabolomics, offers an unprecedented opportunity to move beyond isolated structural insights towards a holistic understanding of biological systems relevant to disease and therapeutic intervention . While AI-predicted protein structures, exemplified by tools like AlphaFold, provide foundational molecular insights, their full potential in drug discovery is unlocked when integrated with the dynamic and comprehensive biological context provided by multi-omics datasets . This integration is crucial for refining target identification, understanding disease mechanisms, and predicting drug responses more accurately, thereby addressing current bottlenecks in drug development. An integrated framework would involve profiling multi-omics data to identify molecular signatures, leveraging these insights for target identification and validation, employing AI for sophisticated protein structure prediction, elucidating structure-function relationships within the omics context, facilitating rational drug design, and informing pre-clinical validation and biomarker discovery .
A second critical direction involves the Synergy with Experimental Techniques and Continued AI Advancements. While AI models have revolutionized protein structure prediction by offering rapid and cost-effective insights, their inherent limitations, particularly in capturing protein dynamics and accurately predicting binding affinities, necessitate a robust hybrid approach . Future advancements in AI are expected to enhance predictive power, but their optimal utility lies in synergistic integration with experimental validation and refinement methods. This includes combining AI predictions with molecular dynamics simulations, quantum mechanics/molecular mechanics (QM/MM) methods, and experimental validation through synthesis and biological assays . The continuous development of more comprehensive AI platforms that can handle diverse, computationally intensive tasks will ensure that AI-driven structural predictions are integral components of an expansive and iterative drug development pipeline .
Finally, the increasing reliance on AI in drug discovery brings forth significant Ethical Considerations and Regulatory Frameworks. Despite the technical focus of much of the current literature, the responsible deployment of AI necessitates a detailed discussion of broader ethical implications, including data privacy, algorithmic bias, and the evolving regulatory landscape . Algorithmic bias, stemming from non-diverse training datasets, poses a risk to fairness and equity in drug development outcomes. Addressing this requires proactive strategies such as diverse data curation, bias detection and mitigation algorithms, enhanced transparency and explainability in AI models, and the establishment of robust regulatory oversight and ethical guidelines . Furthermore, discussions around open access, data sharing, and standardized data formats are crucial for fostering collaboration and ensuring equitable access to powerful AI tools, highlighting the tension between open science principles and proprietary control . The trajectory of AI in drug discovery clearly points to a future where ethical guidelines and regulatory frameworks will be indispensable for ensuring the responsible and equitable application of these transformative technologies.
5.1 Integration of Multi-Omics Data with Structural Biology
The increasing availability of multi-omics data (e.g., genomics, transcriptomics, proteomics, metabolomics) presents a significant opportunity to enhance drug discovery by providing a more holistic understanding of biological systems . While AI-predicted protein structures offer unprecedented insights into molecular targets, integrating these structures with multi-omics data can lead to more comprehensive drug discovery strategies, addressing current bottlenecks in drug development.
The integration of protein structure prediction with multi-omics data can provide a deeper understanding of disease mechanisms and therapeutic interventions. For instance, while several studies focus on AI's role in protein structure prediction for drug discovery, they often do not explicitly detail the integration of multi-omics data as a central theme . However, some research implicitly supports this integration. For example, some studies suggest that refining protein structure predictions can be achieved by incorporating protein sequence information alongside contextual data, such as the protein's environment (other molecules) or experimental findings like X-ray crystallography . This highlights the potential of contextual data, which multi-omics datasets can provide, to enhance the accuracy and relevance of structural predictions.
Furthermore, tools like AlphaFold 3 are posited to offer a "new window on the molecules of life," revealing the intricate connections within cellular systems and their influence on biological functions . This perspective inherently supports the integration of structural insights with broader cellular understanding, which multi-omics data can comprehensively provide. Similarly, the application of tools like PandaOmics for target selection in specific diseases, such as hepatocellular carcinoma (HCC), demonstrates an indirect link to integrating biological context with structural information, even if it does not explicitly detail a multi-omics integration framework . This points towards a developing recognition of the need for contextual biological information to inform structural drug discovery.
To overcome current bottlenecks in drug development, an improved framework leveraging the integration of AI-predicted protein structures with multi-omics data can be proposed. This framework would involve several stages:
- Omics Data Profiling: Comprehensive collection and analysis of multi-omics data (genomics, transcriptomics, proteomics, metabolomics, epigenomics) from diseased and healthy tissues to identify molecular signatures and perturbed pathways. For example, the use of AI in single-cell multi-omics models for understanding cellular heterogeneity in tumors and informing personalized cancer treatments aligns with this initial stage.
- Target Identification and Validation: Using multi-omics data to pinpoint potential drug targets within the perturbed pathways identified in the profiling stage. This would involve identifying key proteins (and their variants) that are causally linked to disease pathology.
- AI-Driven Structure Prediction: Employing advanced AI models to predict the 3D structures of identified protein targets, including their various conformational states and dynamics, especially when influenced by other molecules in their environment .
- Structure-Function Relationship Elucidation with Omics Context: Integrating the predicted protein structures with the multi-omics data to understand how structural changes at the molecular level are manifested in cellular and physiological changes observed in the omics profiles. This step would allow for a deeper understanding of genotype-phenotype relationships and the impact of genomic variants .
- Rational Drug Design and Optimization: Utilizing the integrated structural and omics insights to design small molecules that precisely interact with the identified targets. This step would go beyond simple binding prediction, considering the broader cellular context and potential off-target effects informed by multi-omics data.
- Pre-clinical Validation and Biomarker Discovery: Employing multi-omics data in pre-clinical models to validate drug efficacy, assess toxicity, and identify predictive biomarkers for patient stratification, facilitating personalized medicine approaches.
This integrated framework ensures that drug discovery is not solely reliant on static structural predictions but is enriched by the dynamic and comprehensive biological context provided by multi-omics data. Such an approach can significantly reduce the attrition rate in drug development by identifying more relevant targets, designing more effective and safer compounds, and predicting patient responses more accurately.
5.2 Synergy with Experimental Techniques and Continued AI Advancements
The revolutionary strides in AI-driven protein structure prediction, exemplified by tools like AlphaFold and its successors, have profoundly reshaped the landscape of small molecule drug discovery by offering rapid and cost-effective structural insights . However, to ensure the robustness and reliability of these predictions, a hybrid approach that integrates computational methods with experimental validation remains paramount . This synergy is particularly crucial given the inherent limitations of current AI models, especially in capturing the dynamic nature of proteins and precisely predicting binding affinities .
Despite the power of AI models like AlphaFold and AlphaFold3, they still face challenges in handling complex cases, accurately capturing dynamic transitions, improving time efficiency, and reducing reliance on existing experimental data, such as the Protein Data Bank (PDB) . For instance, AlphaFold2 models, while valuable starting points, often require complementary tools like molecular modeling or protein dynamics simulations to infer protein conformational changes, especially in the absence of observed ligand binding data . The article from The Science Advisory Board further emphasizes the necessity of addressing these limitations, particularly concerning protein dynamics and accurate binding affinity predictions, thereby supporting the integration of predictive tools with experimental validation and refinement methods .
The ongoing advancements in AI and machine learning are expected to further enhance the predictive power and scope of protein structure prediction, but their full potential is realized through synergistic integration. For example, DeepMind Technologies is actively refining AlphaFold's capabilities through collaboration with the scientific community . Future directions in AI development include better handling of protein dynamics and conformational changes, and reduced reliance on homologous templates, as well as the use of AI in fragment-based drug design to accelerate drug identification . Brown University's work, for instance, explores combining subsampled AlphaFold2 with molecular dynamics (MD) simulations to better understand protein motions, such as stretching and bending, and automating this data subsampling for large-scale analysis of protein dynamics . The development of methods to predict multiple protein configurations and their frequencies represents a significant advancement, with future research exploring the integration of this dynamic prediction capability with experimental techniques for validation and refinement .
The synergy between computational and experimental methods is evident in various aspects of drug discovery. Molecular Dynamics (MD) simulations can validate docking results, and Quantum Mechanics/Molecular Mechanics (QM/MM) methods can enhance the accuracy of docking and virtual screening . For de novo drug design, reinforcement learning combined with prediction models can guide generative models for property optimization, establishing a crucial feedback loop where predictions inform generation . An illustrative example of this synergistic workflow is a study that integrated computational predictions from AlphaFold and Chemistry42 with experimental validation through synthesis and biological assays. This iterative process, involving the generation, testing, and subsequent AI-powered refinement of compounds, demonstrated AlphaFold's role in accelerating the discovery of a novel CDK20 small molecule inhibitor .
To specifically address AI prediction shortcomings, integrating tools like AF2RAVE with other computational methods is vital . For example, combining AF2RAVE with ligand binding site predictors (e.g., GrASP) to create comprehensive workflows (AF2RAVE-GrASP-Glide), and incorporating free energy perturbation (FEP) calculations for binding affinity evaluation, significantly improves accuracy and sampling of protein conformations for drug design . Furthermore, the output of tools like AF2BIND, which provides a hierarchy of residues involved in binding, offers richer information than purely volumetric pocket-finding algorithms, allowing for synergistic use where its predictions can refine experimental data or guide further computational analysis . The release of the AlphaFold Server, an easy-to-use research tool, further facilitates this synergy by enabling scientists to quickly generate predictions for hypothesis generation and accelerate experimental workflows . The collaborative efforts between Isomorphic Labs and pharmaceutical companies to apply AlphaFold3 to real-world drug design challenges underscore the importance of this combined approach . The future of drug discovery lies in the continuous development of more comprehensive AI platforms that can handle diverse computationally intensive tasks, ensuring that AI-driven structural predictions are not isolated but rather integral components of an expansive and iterative drug development pipeline .
5.3 Ethical Considerations and Regulatory Frameworks
The increasing reliance on artificial intelligence (AI) in drug discovery introduces a complex array of ethical considerations that necessitate careful attention. While the provided literature extensively covers technical advancements in protein structure prediction and its application in drug discovery, a notable gap exists in the detailed discussion of the broader ethical implications, data privacy concerns, algorithmic bias, and the regulatory landscape for AI-driven drug development .
Despite the general lack of explicit discussion on these ethical issues within the surveyed papers, the responsible deployment of AI in this domain is paramount. Google DeepMind, in the context of AlphaFold models, emphasizes a "science-led approach" and has conducted extensive assessments with domain experts and specialist third parties across biosecurity, research, and industry to understand capabilities and mitigate potential risks . This proactive engagement with the scientific community and policymakers for responsible AI deployment, coupled with making the AlphaFold Server available for non-commercial research, indicates a strategic approach to responsible sharing and risk mitigation . However, the controlled release strategy for crucial model weights, requiring Google's explicit permission for academic use, highlights a balance between scientific and commercial needs, implicitly raising questions about equitable access and control over powerful AI tools .
A critical ethical consideration is algorithmic bias, which can emerge if the datasets used to train AI models are not diverse or representative. In drug discovery, this could manifest as models performing suboptimally for certain demographic groups or disease states if the underlying biological data primarily reflects specific populations or conditions. While the surveyed digests do not explicitly detail specific challenges posed by algorithmic bias in drug discovery pipelines or propose mitigation strategies, this issue is fundamental to ensuring fairness and equity in the application of these advanced technologies . Potential interdisciplinary solutions for fairness and equity in AI-driven drug discovery would involve: (1) Diverse Data Curation: Actively seeking and incorporating data from diverse genetic backgrounds, ethnicities, and disease cohorts to prevent biases in model training. (2) Bias Detection and Mitigation Algorithms: Developing and implementing algorithms capable of detecting and correcting biases within AI models. (3) Transparency and Explainability: Ensuring that AI models are not black boxes, allowing researchers to understand how decisions are made and identify potential biases. (4) Regulatory Oversight and Ethical Guidelines: Establishing robust regulatory frameworks and ethical guidelines that mandate diversity in data, require bias auditing, and promote equitable access to the benefits of AI-driven drug discovery.
To foster collaboration and accelerate progress while addressing these ethical challenges, the importance of open access, data sharing, and standardized data formats cannot be overemphasized . While some models like AlphaFold have free code available under a Creative Commons license, the access restrictions on model weights for AlphaFold3 illustrate the tension between open science principles and proprietary control . Increased openness in data and model access would facilitate independent validation, foster community-driven improvements, and help identify and mitigate biases more effectively. The general trend of AI integration in drug design and ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) prediction, while outside the explicit scope of the current digests, indirectly implies the need for a comprehensive ethical framework. As AI becomes more deeply embedded in these critical stages, the potential for widespread impact on patient populations and healthcare equity underscores the urgency of proactive ethical and regulatory considerations. While the current literature provides limited explicit guidance, the trajectory of AI in drug discovery clearly points to a future where ethical guidelines and regulatory frameworks will be indispensable for ensuring responsible and equitable application of these transformative technologies.
6. Conclusion
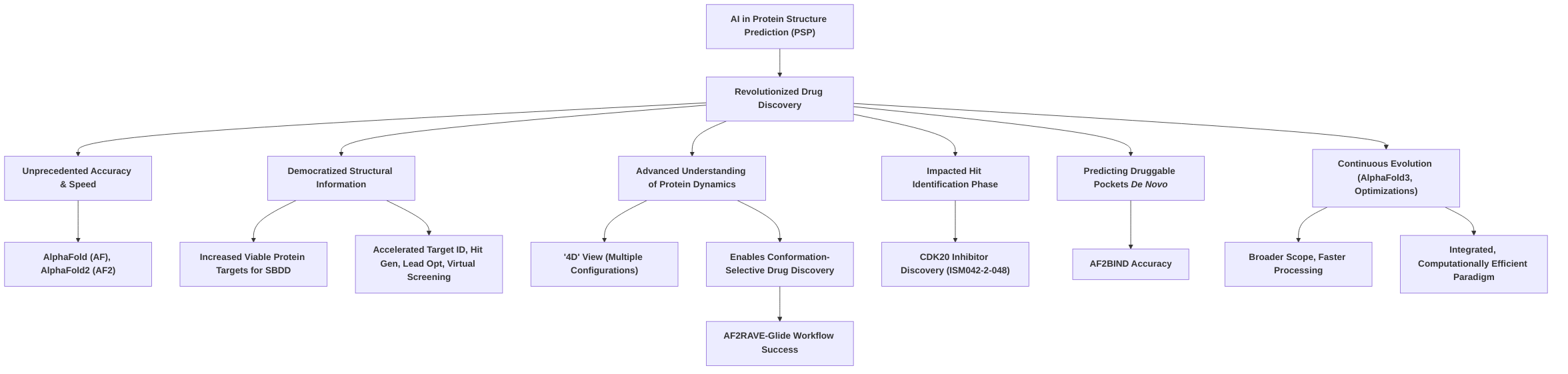
The advent of AI, particularly models like AlphaFold and AlphaFold2, has fundamentally transformed small molecule drug discovery by providing unprecedented accuracy and speed in protein structure prediction . This shift has democratized access to high-resolution protein structures, moving away from resource-intensive experimental methods and thereby accelerating target identification, hit generation, lead optimization, and virtual screening, particularly when leveraging vast compound databases . Beyond static structures, AI has begun to unravel protein dynamics, offering a "4D" view that is critical for understanding conformational changes and enabling the discovery of conformation-selective drugs, as demonstrated by integrated workflows like AF2RAVE-Glide . AI has also proven instrumental in the efficient identification of novel drug candidates for previously uncharacterized targets and in predicting druggable pockets on de novo or predicted structures, showcasing its comprehensive impact across various drug discovery phases . The continuous evolution, exemplified by AlphaFold3's broader scope in predicting molecular interactions and ongoing optimizations for faster processing, underscores a clear trajectory towards an integrated, computationally efficient paradigm for drug discovery .
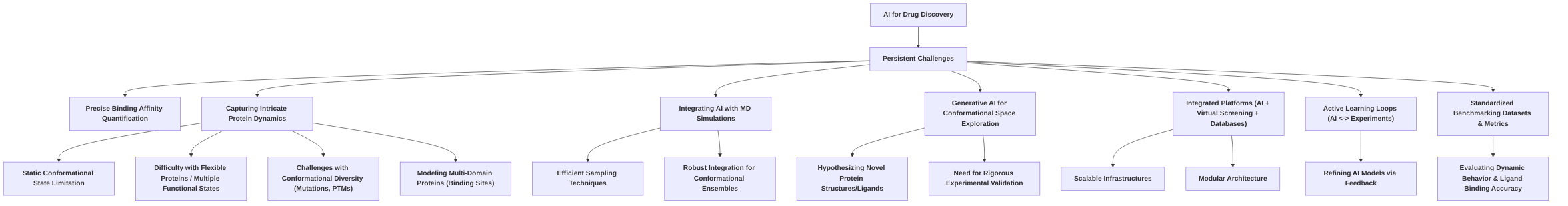
Despite these profound advancements, significant challenges remain. Current AI models often struggle with precisely quantifying binding affinities and fully capturing the intricate dynamic nature of proteins, frequently providing only a static conformational state, which is insufficient for understanding flexible proteins or those undergoing significant conformational changes upon ligand binding . Addressing these limitations necessitates a concerted effort in future research. Key directions include integrating AI with molecular dynamics (MD) simulations to accurately model protein flexibility and conformational ensembles, and leveraging generative AI to explore novel protein structures and ligand designs, while ensuring rigorous experimental validation . The development of integrated platforms that seamlessly combine AI structure prediction with high-throughput virtual screening of massive chemical libraries is crucial . These platforms should foster active learning loops, where computational predictions are continually refined through experimental validation, necessitating close interdisciplinary collaboration . Furthermore, establishing standardized benchmarking datasets and metrics is essential for evaluating the models' ability to predict dynamic protein behavior and ligand binding, ensuring the continued progression towards a more rational and efficient drug design paradigm. The ultimate vision is to move "Beyond AlphaFold2" by developing integrated, experimentally validated computational platforms that overcome existing limitations, thereby accelerating the discovery of groundbreaking therapies.
6.1 Recapitulation of Key Impacts
The advent of artificial intelligence (AI) in protein structure prediction has fundamentally reshaped small molecule drug discovery, ushering in a paradigm shift from conventional, often time-consuming methods to a more efficient, data-driven, and accelerated process . This transformation is primarily driven by the remarkable accuracy and speed achieved by deep learning (DL) models, most notably AlphaFold, which has revolutionized the field by enabling the rapid prediction of intricate protein structures and their dynamic behaviors .
One of the most significant contributions is the democratization of accurate protein structure information. Prior to AI, obtaining high-resolution protein structures was a bottleneck, heavily reliant on experimental techniques such as X-ray crystallography or NMR spectroscopy, which are often costly and technically challenging . AlphaFold2, in particular, has made highly accurate protein structure predictions widely accessible, thereby increasing the number of viable protein targets for structure-based drug design (SBDD) and accelerating crucial stages like hit and lead generation, target identification, and virtual screening . This low-cost prediction capability has also significantly bolstered drug repositioning and virtual screening efforts, especially when coupled with large compound databases like ZINC20, opening new avenues for efficient small molecule drug identification .
Beyond static structure prediction, AI-powered methods have advanced the understanding of protein dynamics. A key innovation is the ability to rapidly predict multiple protein configurations, moving beyond a single static representation to a more comprehensive "4D" view of proteins, which is crucial for capturing the conformational landscape essential for drug discovery . While standard AlphaFold2 predictions may be insufficient for targeting metastable protein conformations, integrated workflows like AF2RAVE-Glide, which combine AlphaFold2 with enhanced sampling and docking, have successfully modeled these critical states. This approach has demonstrated a significant improvement in the success rate of docking type II kinase inhibitors, highlighting its potential for discovering conformation-selective drugs and the transferability of learned latent spaces across homologous kinases .
Furthermore, AI has profoundly impacted the hit identification phase of drug discovery. An illustrative example is the efficient discovery of a potent small molecule inhibitor (ISM042-2-048) for CDK20, a novel target without an experimental structure, through an end-to-end AI-powered workflow. This workflow integrated target selection (PandaOmics) with generative chemistry (Chemistry42), showcasing AlphaFold's direct role in significantly reducing the time and cost associated with drug discovery .
The utility of AlphaFold2 extends to identifying druggable pockets even on previously uncharacterized proteins. AF2BIND, a model trained on AlphaFold2's internal pair representation, can accurately predict small-molecule-binding residues de novo, achieving a 66% recovery. Its interpretability, which allows for correlating "bait" amino acid contributions with ligand hydrophobicity, provides novel insights into binding site characteristics, further accelerating drug discovery by enabling the identification of druggable pockets on novel or predicted structures .
Recent advancements, such as AlphaFold3, represent an expansion of AI's capabilities beyond individual protein structures to include complex molecular interactions with DNA, RNA, and ligands . This broader scope and improved accuracy in predicting molecular interactions are expected to further transform biological understanding and accelerate drug discovery by offering a more complete picture of biological systems. The continued optimization of AlphaFold2 implementations, achieving 22x faster processing times while maintaining accuracy through techniques like split-compute architecture and GPU-accelerated search, underscores the relentless pursuit of efficiency in AI-driven drug discovery . These advancements signify a clear shift towards an integrated, computationally intensive, and highly efficient paradigm for drug discovery.
6.2 Outlook and Unresolved Issues
Despite significant advancements in protein structure prediction, notably by AlphaFold2 and AlphaFold3, several critical limitations persist, particularly concerning their direct applicability to small molecule drug discovery . A primary unresolved issue is the difficulty in precisely quantifying binding affinities and capturing the dynamic nature of proteins . Current AI models often predict only a single, static conformational state, which is insufficient for understanding proteins that undergo significant conformational changes upon ligand binding, exhibit high flexibility, or possess multiple functional states . This limitation extends to predicting conformational diversity arising from mutations or post-translational modifications and accurately modeling multi-domain proteins where ligand binding may involve global domain movement or cryptic binding sites . Furthermore, issues such as significant residue clashes and insufficient exploration of conformational space remain challenging for existing methods .
To address these gaps, future research must focus on developing AI models that explicitly incorporate protein dynamics and learn from comprehensive experimental binding data . One promising direction involves integrating AI structure prediction with molecular dynamics (MD) simulations . While MD simulations can provide detailed insights into protein flexibility and conformational changes, their high computational cost and challenges in validation pose significant hurdles. Specifically, efforts should be directed towards developing efficient sampling techniques for MD and integrating them with AI predictions to generate robust conformational ensembles. For instance, subsampling methods applied to AlphaFold2 could be automated and further integrated with MD simulations to predict protein dynamics more accurately .
Another critical research direction involves leveraging generative AI for exploring conformational spaces and hypothesizing novel protein structures or ligand designs . This approach requires rigorous experimental validation pipelines to confirm the biological relevance of computationally generated structures and ligands. Challenges include ensuring that generative models produce physically plausible and biochemically active molecules, as well as developing robust methods to address and mitigate residue clashes in predicted structures .
The creation of integrated platforms that seamlessly combine optimized AI structure prediction with real-time virtual screening of massive chemical libraries represents a significant interdisciplinary opportunity . Such platforms could adopt a modular architecture, integrating various AI tools for target selection, structure prediction, ligand binding site prediction, and molecule generation . For example, an integrated platform could combine AF2RAVE for conformational sampling with ligand binding site predictors and advanced binding affinity calculation methods like Free Energy Perturbation (FEP) . The feasibility of such platforms hinges on developing highly scalable computational infrastructures and robust data management systems to handle the exponential growth in chemical availability and the demands of high-throughput screening .
Furthermore, establishing active learning loops is crucial, wherein promising computationally predicted drug candidates are prioritized for experimental validation, creating a continuous feedback mechanism to refine and improve AI models . This requires close collaboration between computational biologists, experimental chemists, and structural biologists to ensure efficient feedback mechanisms and robust experimental validation pipelines. The success of these loops relies on accurately identifying and synthesizing high-confidence candidates and swiftly validating their activity and specificity in vitro and in vivo.
Finally, the development of standardized benchmarking datasets and metrics is essential for evaluating the ability of AI models to predict protein conformational ensembles and their relevance to ligand binding . These datasets should include diverse protein families, targets with inherent flexibility, and a range of ligand types. Evaluation criteria should extend beyond static structure prediction accuracy to encompass the quality of predicted conformational states, the ability to identify cryptic binding pockets, and the accuracy of predicted binding affinities for diverse ligands.
In conclusion, the integration of AI-driven protein structure prediction with small molecule drug discovery holds immense potential for accelerating the development of new therapeutics and addressing unmet medical needs. Achieving this vision necessitates a concerted interdisciplinary effort to overcome current limitations in capturing protein dynamics, accurately predicting binding affinities, and creating seamlessly integrated, experimentally validated computational platforms. By focusing on these critical areas, the field can move "Beyond AlphaFold2" to unlock a new era of rational drug design, bringing groundbreaking therapies to patients more rapidly and cost-effectively.
References
Advances in Protein Structure Prediction for Drug Design - Number Analytics https://www.numberanalytics.com/blog/advances-protein-structure-prediction-drug-design
AlphaFold 3 predicts the structure and interactions of all of life's molecules - Google Blog https://blog.google/technology/ai/google-deepmind-isomorphic-alphafold-3-ai-model/
AlphaFold accelerates artificial intelligence powered drug discovery: efficient discovery of a novel CDK20 small molecule inhibitor - Chemical Science (RSC Publishing) https://pubs.rsc.org/en/content/articlelanding/2023/sc/d2sc05709c
The Revolutionary Impact of AlphaFold on Drug Discovery: Decoding the Mystery of Protein Folding - Lindus Health https://www.lindushealth.com/blog/the-revolutionary-impact-of-alphafold-on-drug-discovery-decoding-the-mystery-of-protein-folding
Advances in AI for Protein Structure Prediction: Implications for Cancer Drug Discovery and Development - PMC https://pmc.ncbi.nlm.nih.gov/articles/PMC10968151/
AI Guided Protein Structure Prediction: What does it mean for drug discovery? https://www.tarletoncomms.com/insights/ai-in-drug-development-protein-prediction
Application of Computational Biology and Artificial Intelligence in Drug Design - PMC https://pmc.ncbi.nlm.nih.gov/articles/PMC9658956/
Alphafold2 protein structure prediction : Implications for drug discovery - PMC https://pmc.ncbi.nlm.nih.gov/articles/PMC7614146/
AlphaFold3: Revolutionizing drug discovery and development - Labiotech.eu https://www.labiotech.eu/in-depth/alpha-fold-3-drug-discovery/
Protein structure prediction via deep learning: an in-depth review - Frontiers https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1498662/full
AF2BIND: Predicting ligand-binding sites using the pair representation of AlphaFold2 https://www.biorxiv.org/content/10.1101/2023.10.15.562410v1.full-text
Empowering AlphaFold2 for protein conformation selective drug discovery with AlphaFold2-RAVE - eLife https://elifesciences.org/reviewed-preprints/99702
AF2BIND: Predicting ligand-binding sites using the pair representation of AlphaFold2 - bioRxiv https://www.biorxiv.org/content/10.1101/2023.10.15.562410v1.full.pdf
MIT team finds limitations of AlphaFold's AI protein structures in drug discovery https://www.scienceboard.net/index.aspx?sec=sup\&sub=Drug\&pag=dis\&ItemID=4688
Accelerating Drug Discovery with AlphaFold2 Optimization - Salt AI https://www.salt.ai/blog/accelerating-drug-discovery-with-alphafold2-optimization
Brown University's Subsampled AlphaFold2 Approach for Predicting Protein Dynamics: A Game-changer for Drug Discovery? - CBIRT https://cbirt.net/brown-universitys-subsampled-alphafold2-approach-for-predicting-protein-dynamics-a-game-changer-for-drug-discovery/
AlphaFold2 + ZINC20, open a new era of virtual drug screening! | MedChemExpress https://www.medchemexpress.com/literature/alphafold2-zinc20-open-a-new-era-of-virtual-drug-screening.html
Protein Structure Prediction: A Viable Tool for Drug Design - Chettinad Health City Medical Journal https://www.chcmj.ac.in/backend/images/article\_pdf/jqH9q2qidtvi2geMHgCHukjJZvM4qR5tEIIqIJTj.pdf
New technique for predicting protein dynamics may prove big breakthrough for drug discovery | Brown University https://www.brown.edu/news/2024-03-27/protein-models