0. Unveiling the Tumor Immune Microenvironment with Single-Cell Multi-Omics: A Systematic Review
1. Introduction
The Tumor Immune Microenvironment (TIME) represents a dynamic and complex ecosystem integral to cancer progression, metastasis, and therapeutic response . It comprises various cellular components, including cancer cells, diverse immune and stromal cells, and non-cellular elements like the extracellular matrix, cytokines, and growth factors, all interacting intricately to dictate cancer biology .
Historically, our understanding of TIME was largely derived from bulk analyses, such as bulk RNA-seq, which, despite providing general overviews, suffered from significant limitations. These methods average signals from heterogeneous cell populations, thereby obscuring critical information from rare cell types and failing to resolve the fine-grained heterogeneity within tumors . This deficiency was particularly pronounced in characterizing tumor heterogeneity, encompassing genetic, epigenetic, phenotypic, and functional variations crucial for progression and treatment resistance .

The evolution of TIME research has undergone a significant paradigm shift from reductionist bulk analyses to comprehensive systems biology approaches, driven by continuous technological advancements . While systems biology laid groundwork for understanding complex dynamics, it often still faced challenges in resolving granular cellular details when relying on aggregated data .
The advent of single-cell multi-omics technologies has addressed these limitations by enabling the study of individual cellular behaviors and interactions with unprecedented resolution, overcoming the averaging effect of bulk analyses and providing insights into previously masked heterogeneity and dynamics . These technologies are crucial for resolving cellular heterogeneity, identifying novel cell states and rare populations, and characterizing intricate cell-cell interactions within the TIME . They also excel at revealing dynamic transitions and hybrid cell states, which are pivotal for understanding cancer and immune cell plasticity, drug resistance, and immune evasion mechanisms .
This systematic review aims to comprehensively outline the latest advancements in single-cell multi-omics techniques and their applications in cancer research, with a specific emphasis on the TIME . The objectives include cataloging and comparing relevant single-cell multi-omics technologies, elucidating their role in revealing cancer cell plasticity and intratumoral heterogeneity, characterizing immune cell diversity and interactions within the TIME, and identifying current clinical applications and future directions for advanced diagnostic and therapeutic strategies . The scope will encompass various single-cell multi-omics techniques, with a focus on understanding cancer cell plasticity, intratumoral heterogeneity, cancer cell evolution, and the formation of hybrid cell states within the TIME, while acknowledging the broader field of Cancer Systems Immunology .
1.1 Background of Tumor Immune Microenvironment (TIME)
The tumor immune microenvironment (TIME) is a multifaceted and dynamic ecosystem comprising various cellular and non-cellular components that profoundly influence cancer progression, metastasis, and therapeutic response . Key cellular constituents include cancer cells, diverse immune cells (e.g., T cells, B cells, myeloid cells), and stromal cells, while non-cellular elements encompass the extracellular matrix, cytokines, and growth factors . The intricate interactions among these components dictate crucial aspects of cancer biology, including immune evasion, treatment resistance, and metastatic potential .
Historically, the understanding of the TIME's complexity was largely derived from bulk analyses, such as traditional high-throughput sequencing methods like bulk RNA-seq . While these approaches provided a general overview, they suffered from significant limitations. Specifically, bulk analyses average signals from mixed cell populations, thereby obscuring critical information from rare cell types and failing to resolve the fine-grained heterogeneity inherent within tumors . This limitation was particularly pronounced in characterizing tumor heterogeneity, which encompasses genetic, epigenetic, phenotypic, and functional variations crucial for tumor progression and treatment resistance . Consequently, the nuanced interactions and specific contributions of individual cell types to cancer progression and treatment response remained largely elusive.
The evolution of systems biology, integrating mathematical modeling and computational analyses with experimental data, paved the way for a more quantitative understanding of these complex biological systems . However, the true leap in dissecting the TIME's complexity has emerged with the advent of single-cell technologies. These advanced techniques offer unprecedented resolution, enabling a detailed characterization of individual cellular components and their intricate interactions within the tumor ecosystem, thereby overcoming the averaging effect of bulk analyses and providing insights into the heterogeneity and nuanced dynamics previously masked . This enhanced resolution is fundamental for advancing our understanding of cancer immunity and developing more effective therapeutic strategies.
1.2 Limitations of Traditional Omics in TIME Characterization
Traditional bulk omics approaches, including bulk RNA sequencing and bulk genomics, have been instrumental in cancer biology but possess inherent limitations in comprehensively characterizing the tumor immune microenvironment (TIME) . A primary limitation stems from their reliance on pooled cellular material, which generates an averaged signal across heterogeneous cell populations . This averaging effect obscures critical cell-specific changes and masks signals from rare cell populations, thereby hindering a detailed understanding of the cellular diversity and function within the TIME .
Specifically, bulk omics approaches are unable to resolve the intricate cellular interactions and heterogeneity that are fundamental to tumor immunology and treatment outcomes . This inability to distinguish individual cell states means that valuable information regarding cell-specific gene expression patterns and critical regulatory mechanisms within the complex TIME remains hidden . For instance, the presence and activity of rare immune cell subsets, which may play pivotal roles in anti-tumor immunity or resistance to therapy, cannot be accurately quantified or characterized using bulk methods .
These limitations of bulk omics directly necessitate the development of higher-resolution methods such as single-cell multi-omics. Single-cell technologies are uniquely positioned to address the very questions that bulk methods fail to answer. For example, single-cell multi-omics can characterize previously unidentifiable rare cell populations, providing insights into their unique molecular profiles and functional states within the TIME . Furthermore, these advanced techniques enable the comprehensive mapping of intricate cell-cell communication networks by simultaneously profiling multiple molecular layers (e.g., transcriptome, proteome, epigenome) at single-cell resolution . This capacity to dissect individual cellular states and their interactions is crucial for uncovering the complex regulatory mechanisms governing tumor progression and immune response, which were previously obscured by the averaged signals of bulk omics .
1.3 Significance of Single-Cell Multi-Omics for TIME and Tumor Heterogeneity Research
Single-cell multi-omics technologies represent a transformative leap in dissecting the intricacies of the Tumor Immune Microenvironment (TIME) and tumor heterogeneity, offering an unprecedented resolution that overcomes the inherent limitations of traditional bulk methods . These advancements are crucial for a comprehensive understanding of tumor progression and for informing the development of targeted therapies .
One of the primary benefits of single-cell multi-omics is its ability to resolve cellular heterogeneity at a granular level, which is often obscured in bulk analyses. This capability allows for the identification of novel cell states and rare populations that are critical players in the TIME but undetectable by traditional approaches . Furthermore, these technologies enable the detailed characterization of cell-cell interactions, providing insights into the complex communication networks within the tumor microenvironment that drive cancer development and immune evasion .
Beyond static snapshots, single-cell multi-omics excels at revealing dynamic transitions and hybrid cell states, which are pivotal for understanding the plasticity of cancer cells and immune cells within the TIME . This capacity for unveiling continuous biological processes, rather than average population behaviors, provides a more comprehensive picture of the biological mechanisms at play within tumors . Such insights into cellular plasticity and transitional states are indispensable for comprehending mechanisms of drug resistance and immune evasion, which are often mediated by dynamic shifts in cell phenotypes.
However, the adoption of single-cell multi-omics technologies necessitates a careful consideration of inherent trade-offs, particularly between depth and breadth, and resolution versus throughput. Approaches focusing on "depth" might analyze a limited number of cells but provide extensive molecular information (e.g., genomic, transcriptomic, epigenomic, proteomic) for each cell, offering high-resolution insights into specific cellular programs and interactions. This depth is critical for identifying subtle changes in gene expression or epigenetic modifications that characterize novel cell states or transitional phenotypes. Conversely, "breadth" approaches allow for the analysis of a much larger number of cells, albeit with less molecular detail per cell. While sacrificing some molecular resolution, these methods offer a broader survey of cellular diversity within complex samples, enabling the discovery of rare cell types and global shifts in cellular composition across different disease stages or treatment responses.
The choice between these approaches directly impacts the understanding of the TIME. High-resolution, low-throughput methods can elucidate the intricate molecular mechanisms underpinning specific immune cell exhaustion pathways or cancer cell adaptation strategies. Conversely, high-throughput, lower-resolution methods are invaluable for mapping the overall cellular landscape of a tumor, identifying shifts in immune infiltrate composition, or assessing heterogeneity across large patient cohorts. The strategic integration of both types of approaches, where broad surveys inform targeted deep dives, is often necessary to achieve a holistic understanding of the TIME and tumor heterogeneity, ensuring that both the broad cellular context and the fine molecular details are captured.
1.4 Survey Objectives and Scope
This systematic review aims to comprehensively outline the latest advancements in single-cell multi-omics techniques and their applications within cancer research, with a specific emphasis on the tumor immune microenvironment (TIME) . The primary objectives of this survey are multifaceted: firstly, to systematically catalogue and compare various single-cell multi-omics technologies relevant for TIME analysis . Secondly, the review seeks to elucidate how these technologies are instrumental in revealing cancer cell plasticity and intratumoral heterogeneity, which are critical drivers of tumor evolution and treatment resistance . Thirdly, a key objective is to characterize the intricate diversity, states, and interactions of immune cells within the TIME, providing insights into the complex interplay between the tumor and the immune system . Finally, the survey aims to identify the current clinical applications and future directions of single-cell multi-omics in TIME research, paving the way for advanced diagnostic and therapeutic strategies .
The scope of this systematic review is meticulously defined to ensure a focused and manageable exploration of the field. It encompasses a broad range of single-cell multi-omics techniques, including but not limited to single-cell RNA sequencing (scRNA-seq), single-cell DNA sequencing (scDNA-seq), single-cell proteomics, and single-cell epigenomics, as applied to cancer research . The analysis will specifically emphasize understanding general cancer cell plasticity, intratumoral heterogeneity, cancer cell evolution, and the formation of hybrid cell states within the TIME . While acknowledging the broader field of Cancer Systems Immunology, which leverages advanced technologies and modeling to understand tumor-immune interactions , this review will specifically concentrate on the experimental and analytical capabilities of single-cell multi-omics in dissecting the TIME. This focused approach ensures a deep dive into the current landscape of single-cell methodologies and their profound implications for unveiling the complexities of tumor immunity and guiding precision therapeutic development.
1.5 Evolution of TIME Research: From Bulk to Systems Approaches
The trajectory of research into the Tumor Immune Microenvironment (TIME) has undergone a significant paradigm shift, evolving from reductionist approaches to comprehensive systems biology methodologies . This progression has been critically driven by continuous technological advancements, which have facilitated a deeper exploration of the TIME's inherent complexity and emergent properties .
Initially, TIME research relied heavily on bulk analyses, which, while capable of classifying molecular subtypes and broadly exploring tumor heterogeneity using early high-throughput sequencing techniques, suffered from a critical limitation: the inability to provide a high-resolution cellular landscape . These bulk methods averaged signals across heterogeneous cell populations, thereby obscuring the distinct behaviors and interactions of individual cellular components within the TIME. Consequently, the complex, multiscale, and temporal elements governing tumor-immune responses could not be fully discerned or understood by analyzing individual components in isolation .
The subsequent adoption of systems biology approaches marked a pivotal advancement. Systems biology, by its very nature, emphasizes the interconnectedness of biological components and aims to appreciate complex emergent behaviors that arise from these interactions . This holistic framework, which integrates diverse data types and often employs mathematical modeling, laid the essential groundwork for comprehending the intricate dynamics of the TIME beyond simple, isolated observations . However, even systems biology, when relying on aggregated data, still faced challenges in fully resolving the granular details of cellular composition and molecular features at the single-cell level. The "emergent properties" highlighted by systems biology necessitated a more granular understanding that bulk methods could not provide .
This persistent gap in understanding, particularly concerning cellular heterogeneity and individual cellular behaviors, underscored the necessity for further technological innovation. The advent of single-cell multi-omics technologies has directly addressed these limitations by enabling the study of individual cellular behaviors and interactions with unprecedented resolution . Single-cell multi-omics is specifically designed to overcome the averaging effect of bulk analyses and the inherent limitations of systems biology approaches that did not fully account for single-cell variability. By providing a deep understanding of cellular compositions and molecular features at the single-cell level, these techniques pave the way for true systems-level insights into the TIME, allowing for a comprehensive appreciation of the overall tumor ecosystem and informing the development of more precise therapeutic strategies .
2. Overview of Single-Cell Multi-Omics Technologies
Single-cell multi-omics technologies represent a paradigm shift in understanding complex biological systems, particularly the tumor immune microenvironment (TIME), by providing unprecedented resolution at the individual cell level.
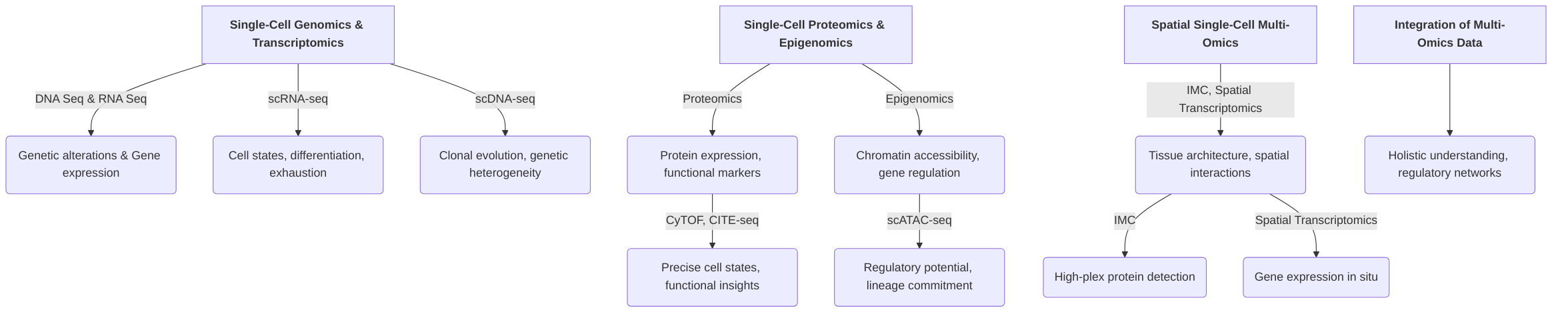
This section outlines the foundational and emerging technologies that enable the comprehensive characterization of cellular states and interactions within the TIME. It begins by detailing single-cell genomics and transcriptomics, including single-cell DNA sequencing (scDNA-seq) and single-cell RNA sequencing (scRNA-seq), which offer insights into genetic alterations and gene expression dynamics, respectively . Subsequently, the discussion moves to single-cell proteomics and epigenomics, highlighting techniques like mass cytometry (CyTOF), CITE-seq, and scATAC-seq, which elucidate protein expression and chromatin accessibility, providing crucial functional and regulatory information that complements genomic and transcriptomic data . The importance of spatial context is then addressed through an overview of spatial single-cell multi-omics technologies, such as Imaging Mass Cytometry (IMC) and Spatial Transcriptomics (ST), which preserve tissue architecture and map molecular profiles in their native spatial coordinates, crucial for understanding cell-cell interactions within the TIME .
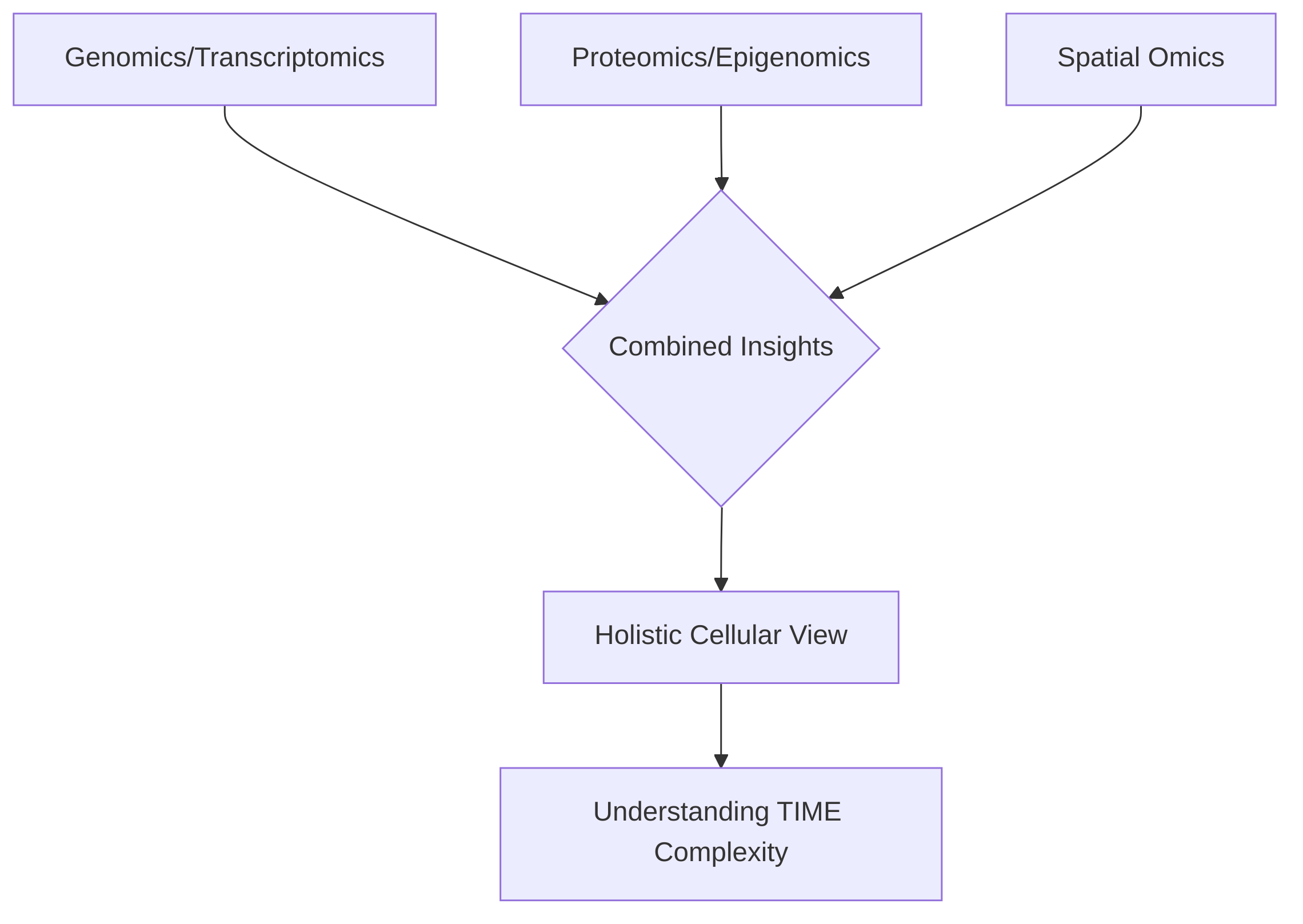
Finally, the section emphasizes the critical role of integrating these diverse single-cell multi-omics datasets. This integration is essential for overcoming the limitations of single-omic analyses, enabling a holistic understanding of cellular heterogeneity, regulatory networks, and dynamic processes within the TIME, despite the inherent computational complexities and challenges in data sparsity and batch effect mitigation .
2.1 Single-Cell Genomics and Transcriptomics
Single-cell DNA sequencing (scDNA-seq) and single-cell RNA sequencing (scRNA-seq) are foundational technologies within the single-cell multi-omics landscape, providing distinct yet complementary insights into the tumor immune microenvironment (TIME) . Both technologies have adapted bulk sequencing principles to achieve single-cell resolution, often leveraging platforms such as 10X Genomics and Smart-seq3 .
scDNA-seq is primarily employed for DNA sequence profiling, enabling the identification of genomic alterations, including single nucleotide variations (SNVs), copy number variations (CNVs), and structural variants (SVs) at the single-cell level . This capability is crucial for investigating the genetic heterogeneity within tumors, as demonstrated by studies analyzing CNA patterns in gastroesophageal junction cancer, which revealed extensive intra-tumor heterogeneity in both primary and metastatic sites . The strength of scDNA-seq lies in its ability to map clonal evolution and understand the genetic drivers of cancer cell plasticity and tumor heterogeneity, offering a stable blueprint of the cell's genetic makeup .
In contrast, scRNA-seq profiles gene expression, providing dynamic insights into cellular states, functions, and transcriptional landscapes . Its application in TIME analysis has been extensive, revealing the cellular and molecular complexity of the immune landscape with unprecedented resolution . For instance, scRNA-seq has been instrumental in characterizing tumor-infiltrating lymphocytes (TILs), particularly cytotoxic CD8+ T cells, elucidating their dysfunctional and exhausted states and identifying a continuum of cell states rather than discrete subtypes within the TME . Furthermore, scRNA-seq aids in understanding the diversity, states, and functions of CD4+ T cell subsets (helper and regulatory) and myeloid cells like dendritic cells and macrophages within the TME . The strength of scRNA-seq lies in its capacity to capture the dynamic transcriptional responses of immune cells and delineate functional heterogeneity within various cancer cell subpopulations .

While scDNA-seq provides information on stable genomic alterations, scRNA-seq offers insights into transient gene expression dynamics, which are crucial for understanding cellular phenotypes and functional states.

The trade-off between sequencing depth and cell number is a critical consideration for both technologies in TIME research. For scDNA-seq, deep sequencing of a smaller number of cells can precisely identify rare mutations and structural variants, which might be missed with lower depth across more cells. However, this limits the detection of rare cell clones or the full extent of genomic heterogeneity across the entire tumor. Conversely, analyzing a high number of cells at lower depth may provide a broader overview of major genomic alterations and clonal lineages but may lack the resolution to identify specific mutations within those lineages. For scRNA-seq, deeper sequencing per cell provides a more comprehensive transcriptome, capturing low-abundance transcripts and fine-tuning cell state definitions. However, this comes at the expense of fewer cells analyzed, potentially missing rare immune cell populations or the full spectrum of heterogeneity in a highly diverse TIME. Conversely, profiling a large number of cells at a shallower depth allows for the detection of more cell types and states, aiding in the discovery of rare populations and broad changes in the immune landscape, but may not fully resolve subtle transcriptional differences or gene regulatory networks. Therefore, the choice between sequencing depth and cell number depends on the specific research question, balancing the need for comprehensive molecular detail against the desire for broad cellular coverage within the complex TIME.
2.2 Single-Cell Proteomics and Epigenomics
Single-cell proteomics and epigenomics offer invaluable insights into the Tumor Immune Microenvironment (TIME) by characterizing cellular states and regulatory mechanisms at an unprecedented resolution, complementing the information derived from genomic and transcriptomic analyses. Single-cell proteomics, employing techniques such as mass cytometry (CyTOF) and Cellular Indexing of Transcriptomes and Epitopes by sequencing (CITE-seq), quantifies protein expression at the single-cell level . These methods are crucial for revealing functional markers and precise cell states, which often are not fully captured by mRNA levels due to post-transcriptional and post-translational modifications . For instance, CyTOF analysis has been instrumental in classifying epithelial cells into distinct subgroups based on proliferation and migration characteristics, providing insights into endocrine resistance mechanisms . CITE-seq further enhances this by concurrently measuring surface protein expression and transcriptomes, allowing for comprehensive characterization of cellular epitopes alongside gene expression profiles .
Conversely, single-cell epigenomics delves into the regulatory landscape of the genome, investigating chromatin accessibility, DNA methylation, and histone modifications. Key techniques include single-cell Assay for Transposase Accessible Chromatin using sequencing (scATAC-seq) and single-cell Chromatin Immunoprecipitation sequencing (scChIP-seq) . These methods offer insights into regulatory potential, gene regulation, and lineage commitment by identifying regions of open chromatin or specific histone marks that indicate active or repressed transcriptional states . For example, scATAC-seq has been effectively used to dissect tumor heterogeneity in glioblastoma and study dynamic evolution in lung adenocarcinoma models, revealing epigenetic features that drive tumor progression .
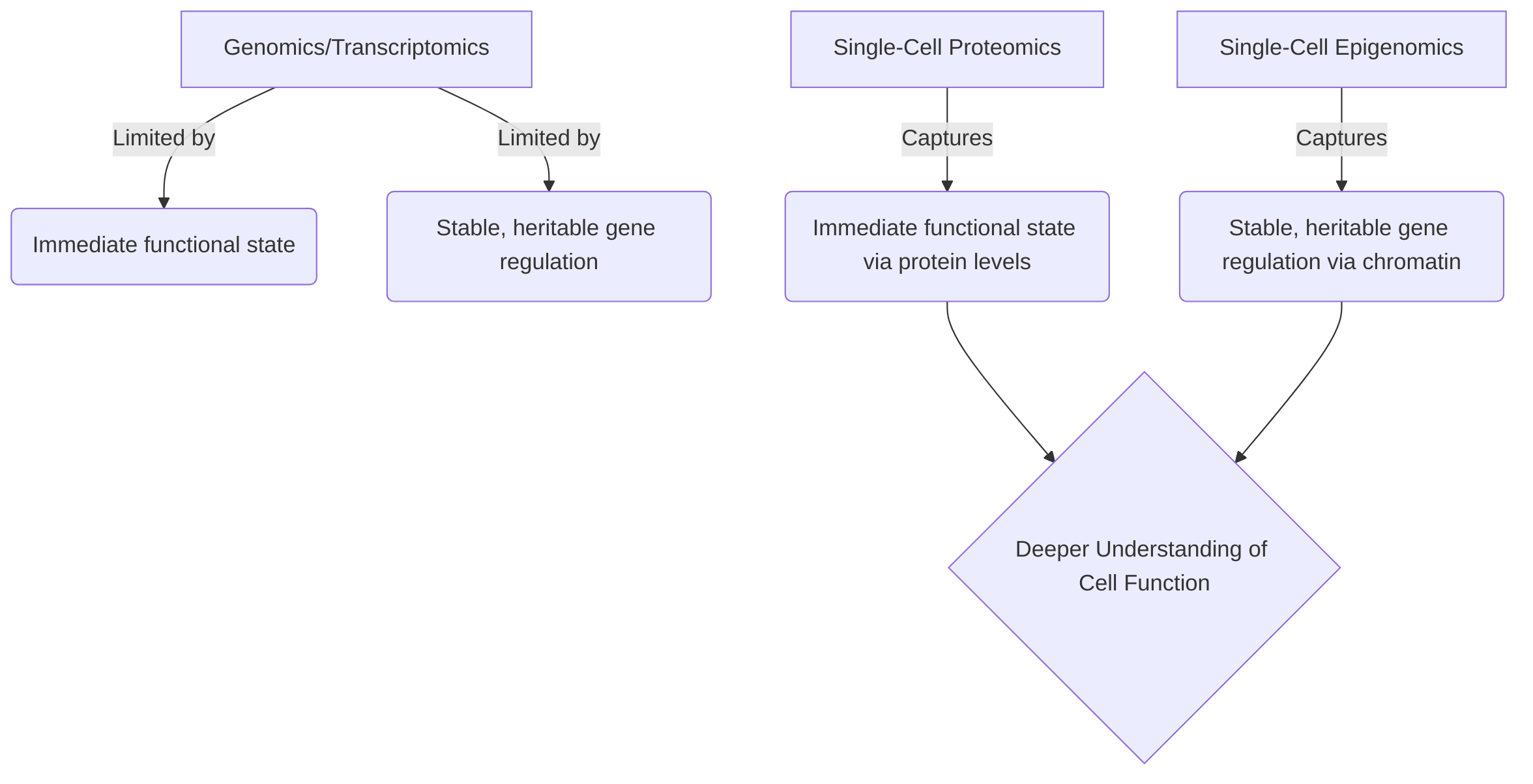
While genomic and transcriptomic approaches provide a foundational understanding of genetic variations and gene expression, they often fall short in capturing the immediate functional state of cells or the stable, heritable changes in gene regulation. Proteomics directly measures the abundance of functional molecules, reflecting the true activity and signaling pathways within a cell. This is critical because protein levels do not always correlate directly with mRNA levels, due to complex post-transcriptional and post-translational regulatory mechanisms . Epigenomics, on the other hand, reveals the potential for gene expression and cellular lineage, explaining how cells with identical genetic material can adopt diverse phenotypes and functions. These epigenetic marks, such as DNA methylation and histone modifications, are dynamic and responsive to environmental cues, providing a layer of regulatory information that dictates cellular identity and adaptability within the TIME .
The strengths of single-cell proteomics lie in its ability to directly quantify functional markers and resolve complex cellular states, which is paramount for identifying therapeutic targets and biomarkers in the TIME. However, a limitation is the current challenges in high-throughput quantification of a broad range of proteins across all cellular compartments, particularly for low-abundance proteins. Single-cell epigenomics excels at mapping gene regulatory landscapes and understanding cell fate decisions, offering insights into long-term cellular memory and plasticity. Its primary limitation pertains to the inherent complexity of epigenetic modifications and the computational challenges in integrating diverse epigenetic datasets to build comprehensive regulatory models.
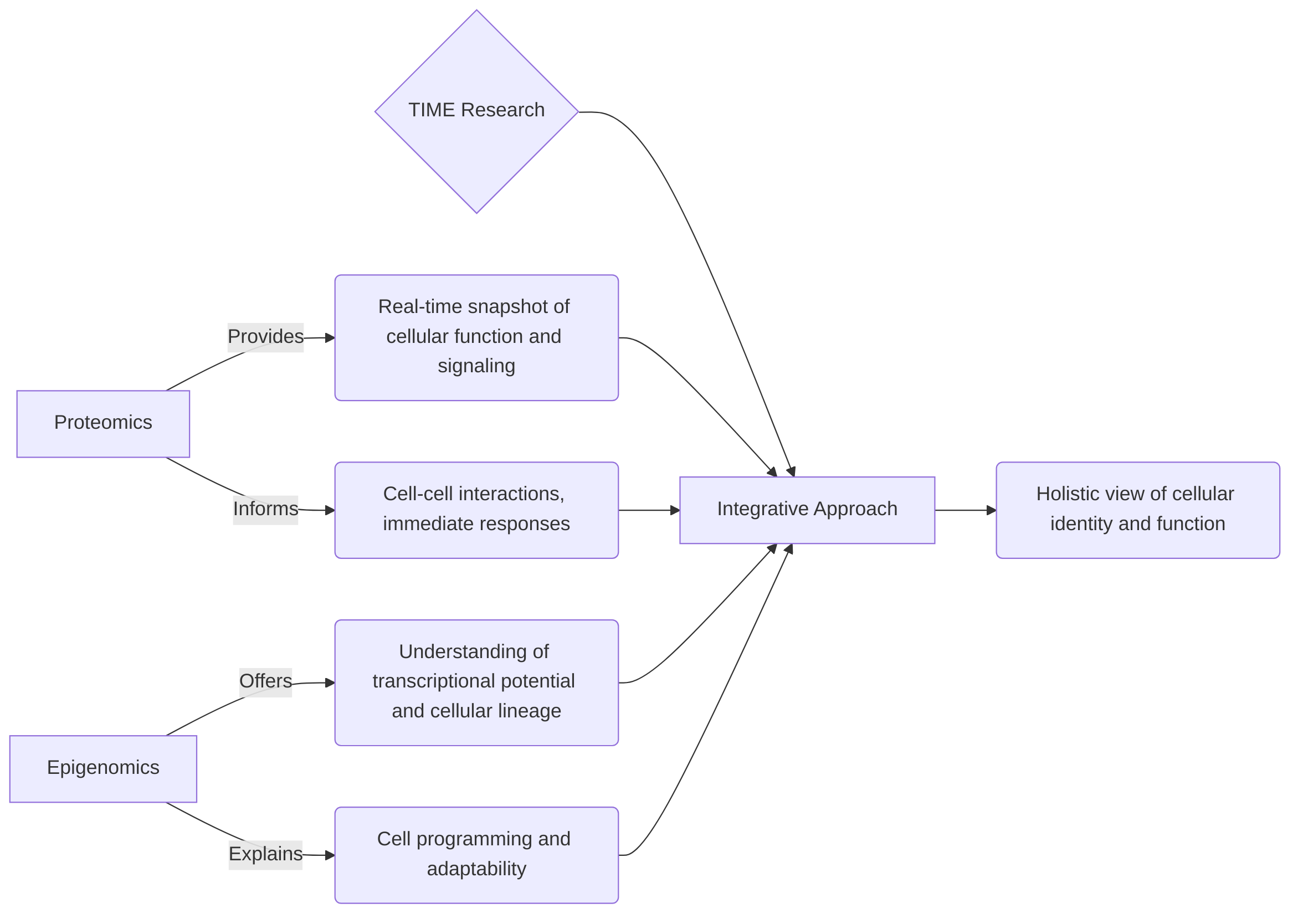
The distinct insights from proteomics and epigenomics are highly complementary for a comprehensive view of the TIME. Proteomics provides a real-time snapshot of cellular function and signaling, directly informing about cell-cell interactions and immediate responses to stimuli. Epigenomics, conversely, offers a deeper understanding of the transcriptional potential and cellular lineage, explaining how cells are programmed to behave. For instance, understanding a cell's lineage commitment through epigenomics can predict its likely protein expression profile, which can then be validated and refined by proteomic analysis. Conversely, an observed protein signature might prompt an investigation into the underlying epigenetic changes driving that particular functional state. By integrating these modalities, researchers can build sophisticated systems-level models that link gene regulatory programs to functional protein outputs, providing a holistic understanding of cellular identity, function, and heterogeneity within the dynamic TIME . Such integrated approaches are crucial for dissecting tumor heterogeneity and informing the development of precision therapeutics .
2.3 Spatial Single-Cell Multi-Omics
Understanding the spatial organization of cells and their interactions within the tumor immune microenvironment (TIME) is crucial for deciphering tumor progression and resistance mechanisms .
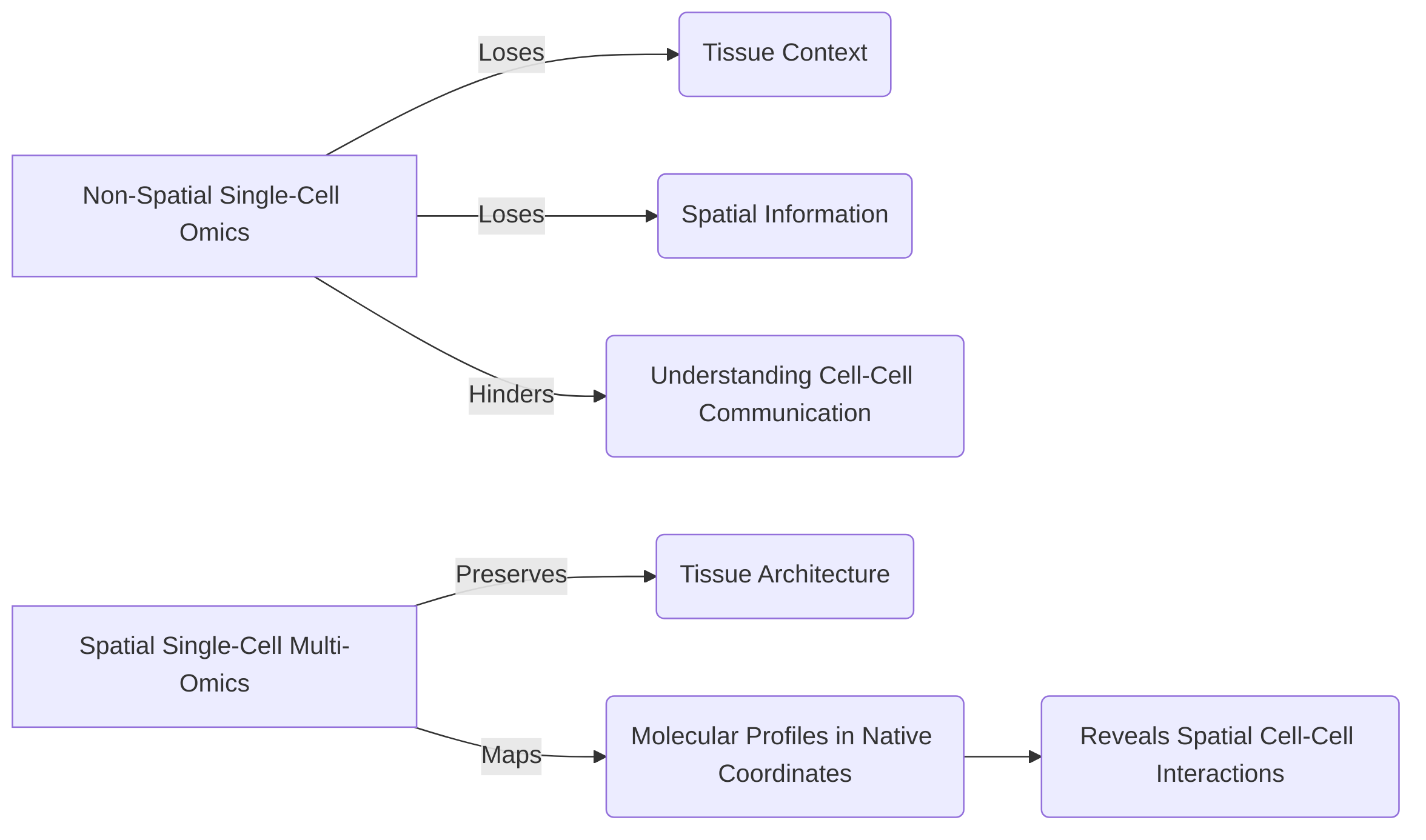
Traditional non-spatial single-cell omics, while providing rich molecular profiles of individual cells, often dissociate cells from their native tissue context, thereby losing critical spatial information about cell-cell communication and tissue architecture . Spatial single-cell multi-omics technologies address this limitation by preserving tissue architecture and mapping cellular and molecular profiles within their original spatial coordinates, offering a more holistic view of the TIME .
Various spatial multi-omics platforms offer distinct capabilities for revealing TIME vulnerabilities. Imaging Mass Cytometry (IMC), for instance, allows for highly multiplexed protein detection, simultaneously measuring over 40 parameters at subcellular resolution on histology slides by using metal-tagged antibodies . This capability is invaluable for identifying and characterizing diverse immune cell populations and their spatial relationships within the tumor, contributing to the understanding of cellular neighborhoods and their impact on immune responses . Similarly, cyclic immunofluorescence techniques like CODEX and multiplexed ion beam imaging (MIBI) provide high-dimensional protein data while preserving spatial context, facilitating the analysis of complex cell-cell interactions and the identification of potential imaging biomarkers .
Spatial Transcriptomics (ST) and related technologies such as seqFISH+ and Slide-seq, on the other hand, focus on profiling gene expression within intact tissue sections . These methods enable researchers to understand the spatial distribution of gene expression patterns and how they correlate with cellular phenotypes and tissue organization. For instance, seqFISH+ offers high spatial resolution and multiplexing capabilities for RNA detection, while Slide-seq combines spatial barcoding with bead-based sequencing to map transcripts spatially . These transcriptomic approaches complement protein-based imaging by providing insights into the transcriptional programs active in specific spatial regions, which can inform about cell states, signaling pathways, and immune evasion mechanisms.
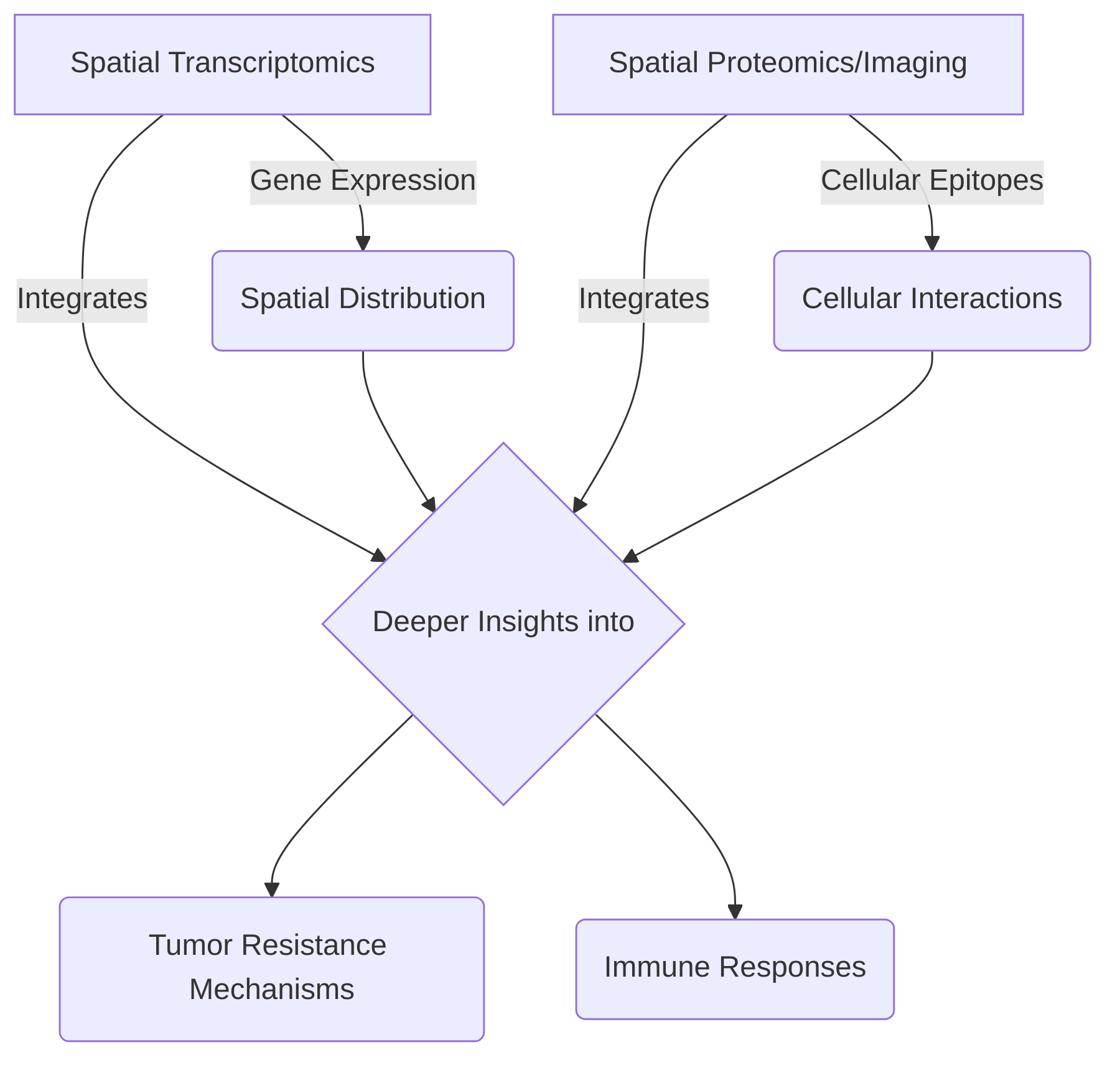
A key advantage of spatial multi-omics platforms is their ability to integrate different modalities of data, such as transcriptomic features, cellular epitopes, and spatial information, to provide deeper insights into tumor resistance mechanisms and immune responses . The PhenoCycler tissue imaging platform, for example, has been utilized for 40-plex longitudinal multimodal single-cell analysis of tumors, integrating various data types to elucidate tumor resistance to immunotherapy . This integration allows for the analysis of cellular neighborhoods and spatial organization, which are critical for understanding how immune cell behavior and communication are influenced by their immediate environment .
Despite their strengths, spatial multi-omics technologies present distinct trade-offs. The primary limitation often lies in the balance between spatial resolution, the number of analytes measured, and throughput. For instance, methods like IMC and MIBI excel in high-plex protein detection at subcellular resolution but may have more limited throughput compared to some spatial transcriptomics approaches . Conversely, Spatial Transcriptomics offers comprehensive gene expression profiles but may have lower cellular resolution depending on the platform. The choice of technology is thus dictated by the specific biological question; high-resolution imaging techniques are ideal for discerning fine-grained cell-cell interactions and sub-cellular localization of molecules, while spatial transcriptomics is more suited for understanding global gene expression changes across tissue regions.
Spatial omics technologies are not merely replacements for non-spatial methods but rather crucial complements. Non-spatial single-cell RNA sequencing (scRNA-seq) provides unparalleled depth in single-cell transcriptomic profiling, enabling the discovery of rare cell types and detailed cell state characterization, which spatial methods may struggle with due to resolution or throughput limitations. However, scRNA-seq loses spatial context. Integrating data from both spatial and non-spatial omics allows researchers to first identify novel cell types and states using scRNA-seq and then map their spatial distribution and interactions within the tissue using spatial multi-omics. This integrated approach provides a comprehensive view of the TIME, bridging the gap between cellular heterogeneity and tissue architecture, ultimately enhancing the understanding of tumor biology and facilitating the identification of novel therapeutic targets. While live-cell imaging and protein detection can be integrated with single-cell multi-omics, specific spatial technologies like ST or IMC are not always extensively detailed in broader reviews, indicating an ongoing need for focused application and development .
2.4 Integration of Single-Cell Multi-Omics Data
The integration of multi-modal single-cell data is paramount for achieving a comprehensive understanding of cellular biology, particularly within complex systems such as the Tumor Immune Microenvironment (TIME). This approach transcends the limitations of single-omic analyses by providing a holistic cellular view, encompassing diverse molecular layers .
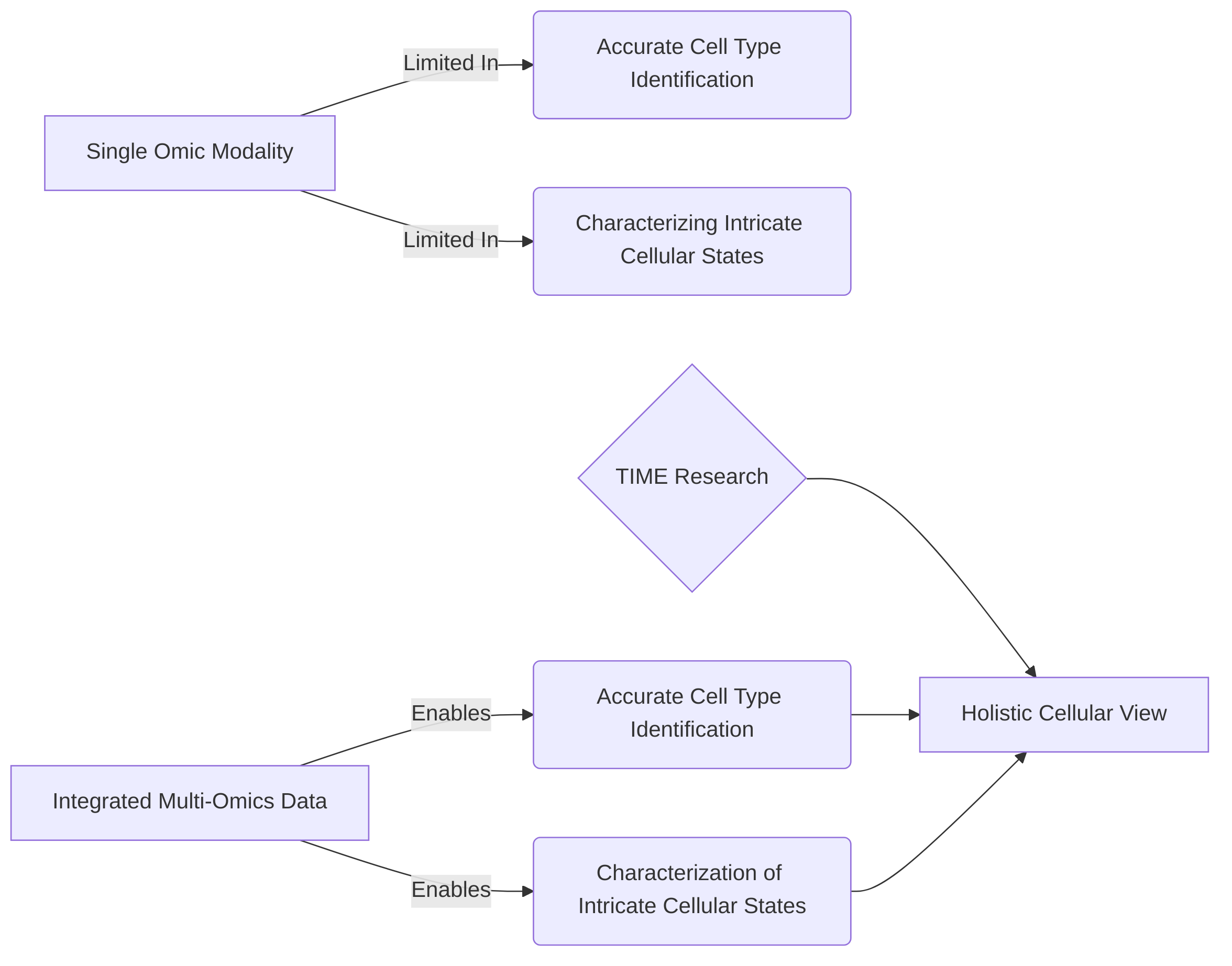
The primary rationale for data integration stems from the need to accurately identify cell types and characterize intricate cellular states, which are often not fully discernible from a single omic modality . By combining diverse data types, such as transcriptomics (RNA-seq), epigenomics (ATAC-seq for histone modifications), and proteomics, researchers can gain deeper insights into cellular differentiation paths, gene regulation, intercellular interactions, spatial organization, cell lineage, and clonal dynamics . For instance, combining RNA-seq with Hi-C data allows for mapping enhancer-gene interactions, thereby providing a more complete picture of transcriptional regulation . A "multimodal integration toolkit" can align transcriptomic features, cellular epitopes, and spatial information, leading to deeper insights into tumor characteristics and responses to therapy .
Integrated analysis offers several key benefits. Firstly, it significantly improves cell type identification and allows for the characterization of complex cellular states that might be ambiguous when viewed through a single molecular lens . Secondly, it facilitates a comprehensive understanding of regulatory networks within the TIME by revealing the interplay between gene expression, chromatin accessibility, and protein abundance . This holistic view enables robust biological discoveries from multi-modal single-cell data, aiding in the interpretation of complex cellular landscapes and the inference of cell state trajectories .
Despite its significant advantages, integrating diverse single-cell omics datasets presents considerable computational complexity. Challenges include handling data sparsity, which is inherent in single-cell data, and mitigating batch effects that can arise from variations in experimental procedures across different datasets . To address these challenges, various methodologies and computational tools are under continuous development. These include weighted-nearest neighbor approaches, multi-view machine learning, and deep generative models . Tools like MAESTRO and Signac are frequently employed to facilitate such analyses, while MaxFuse is emerging as a method for cross-modal data integration .
Key trade-offs exist between different integration approaches. While some methods might excel at aligning distinct data types, others might prioritize preserving biological variability or minimizing computational burden. For instance, multi-level or hybrid analyses can combine continuous models (e.g., Ordinary Differential Equations, Partial Differential Equations) with discrete models (e.g., Agent-Based Models, Cellular Automata) to bridge scales and model cell-cell interactions for a systems-level understanding, though the specific algorithms are not always detailed . The choice of integration method often depends on the specific biological question, the types of data being integrated, and the computational resources available. Therefore, ongoing advancements in computational biology are crucial for developing more efficient and robust integration strategies to unlock the full potential of single-cell multi-omics data in cancer research.
3. Unveiling Immune Cell Diversity and Function in the TIME
The tumor immune microenvironment (TIME) is a complex ecosystem where diverse immune cells interact with cancer cells and stromal components, profoundly influencing tumor progression and response to therapy.
Single-cell multi-omics technologies have emerged as transformative tools, providing unprecedented resolution to dissect the heterogeneity of immune cell populations, decipher their intricate interactions, and understand their dynamic roles in shaping the TIME . This section synthesizes key advancements in utilizing these high-resolution approaches to characterize immune cell diversity, map cellular communication networks, and elucidate the mechanisms underlying response and resistance to immune checkpoint blockade (ICB).
The initial focus involves Characterizing Immune Cell States and Subtypes, where single-cell multi-omics, particularly scRNA-seq, has enabled the precise identification and functional annotation of various immune cell populations, including T cells, B cells, dendritic cells, and macrophages across a wide range of cancers . These technologies have moved beyond traditional classifications, revealing novel cell states, differentiation trajectories, and exhaustion profiles, as exemplified by the continuum of dysfunctional CD8+ T cells or the complex activation states of tumor-associated macrophages (TAMs) . The integration of epigenomic data, such as single-cell ATAC-seq, further enriches this understanding by connecting gene expression patterns to underlying chromatin accessibility and stable functional programs, offering insights into lineage commitment and long-term cellular identity . This deep characterization unveils previously unrecognized immune subsets with prognostic implications, contributing significantly to a comprehensive understanding of immune cell roles within the TIME .
Building upon the identification of individual cell types, the subsequent sub-section, Deciphering Immune Cell Interactions within the TIME, delves into the complex communication networks governing immune responses. Single-cell multi-omics approaches are crucial for mapping cell-cell communication, including chemokine receptor-ligand interactions and spatial relationships, which are vital for comprehending immune cell behaviors and developing effective immunotherapies . Specific examples illustrate how CD8+ T cells recruit other immune cells via CXCL13, how distinct DC subsets orchestrate T cell priming and activation, and how stromal cells, such as cancer-associated fibroblasts (CAFs), influence immunotherapy outcomes through intricate crosstalk with immune cells . A critical aspect highlighted is the integration of spatial multi-omics to move beyond mere correlation and infer causality in cell-cell interactions, providing direct evidence of physical proximity and its impact on immune cell function and communication within defined niches .
Finally, Understanding Immune Checkpoint Blockade Response and Resistance leverages single-cell multi-omics to gain high-resolution insights into the dynamic changes within the immune landscape during ICB treatment. This is pivotal for identifying predictive biomarkers of patient response and uncovering mechanisms of resistance . Key findings include the identification of specific T cell and monocyte subsets that correlate with ICB responsiveness, as well as the revelation of resistance mechanisms such as the limited reinvigoration capacity of dysfunctional TILs, CD58-mediated resistance, and the influence of specific tumor subclones or stromal cell interactions . While the utility of single-cell multi-omics in this context is substantial, continuous efforts are needed to ensure the robustness and reproducibility of these predictions and to biologically validate the identified mechanisms in larger cohorts and preclinical models, thereby paving the way for more precise and effective immunotherapeutic strategies .
3.1 Characterizing Immune Cell States and Subtypes
Single-cell multi-omics technologies have revolutionized the characterization of diverse immune cell populations within the tumor immune microenvironment (TIME), offering unprecedented resolution into their states, subtypes, and functional roles . These approaches are broadly applicable across various cancer types, including hematologic malignancies (e.g., Multiple Myeloma, Lymphoma) and numerous solid tumors (e.g., CRC, Ovarian Cancer, Lung Carcinoma) .
Single-cell RNA sequencing (scRNA-seq) has been a primary tool for dissecting immune cell heterogeneity, enabling the identification of distinct cell types, activation states, differentiation trajectories, and exhaustion profiles . For instance, scRNA-seq has revealed the progression of CD8+ T cells from an effector state to a dysfunctional state in melanoma, highlighting that these dysfunctional cells are often highly clonal and proliferating within the tumor microenvironment . This mirrors a broader finding that intratumoral CD8+ T cells frequently exist in a continuum of dysfunctional and exhausted states . Beyond T cells, scRNA-seq has also elucidated the diverse populations within myeloid cells, such as dendritic cells (e.g., cDC1, cDC2, pDCs) and macrophages (e.g., tumor-associated macrophages, monocytes, MDSCs), uncovering their unique functional states and differentiation pathways . Systems immunology approaches, bolstered by scRNA-seq and mass cytometry, have challenged the conventional M1/M2 polarization model for macrophages, demonstrating a more complex spectrum of activation states and mixed phenotypes among tumor-associated macrophages (TAMs) in various cancers . Similarly, the heterogeneity of dendritic cell subsets and their roles in antigen presentation have been clarified through unbiased single-cell analyses .
The integration of multiple omics layers provides a more holistic view of immune cell biology. While transcriptomics (scRNA-seq) excels at defining activation states and immediate functional repertoires, epigenomics (e.g., single-cell ATAC-seq) provides insights into cell lineage, differentiation potential, and stable functional programs by revealing chromatin accessibility and regulatory landscapes . For example, epigenetic profiling has demonstrated that T cell exhaustion states are epigenetically regulated, with distinct chromatin states differentiating recoverable dysfunctional T cells from those with irreversible dysfunction . This multi-layered approach allows for a deeper understanding of how gene expression patterns (transcriptomics) are influenced by underlying regulatory mechanisms (epigenomics), contributing to the long-term identity and behavior of immune cells.
Furthermore, single-cell multi-omics has identified previously unrecognized immune cell subsets and their specific roles in anti-tumor immunity . For instance, the presence of peri-tumoral lymphoid aggregates, enriched with B cell-associated signatures, has been linked to improved patient survival in certain contexts, indicative of an "immune-striving" tumor microenvironment . This highlights how high-resolution single-cell analysis can uncover specific immune cell constellations and their prognostic implications, thereby contributing significantly to understanding the diverse roles of immune cells, including T cells, B cells, macrophages, and dendritic cells, in shaping the complex landscape of the TIME .
3.2 Deciphering Immune Cell Interactions within the TIME
Single-cell multi-omics technologies have significantly advanced our understanding of the intricate communication networks among immune cells and other components within the tumor immune microenvironment (TIME) . These approaches enable the analysis of cellular composition, chemokine receptor-ligand networks, and spatial distribution, providing a detailed map of cellular crosstalk essential for comprehending immune responses and developing effective immunotherapies .
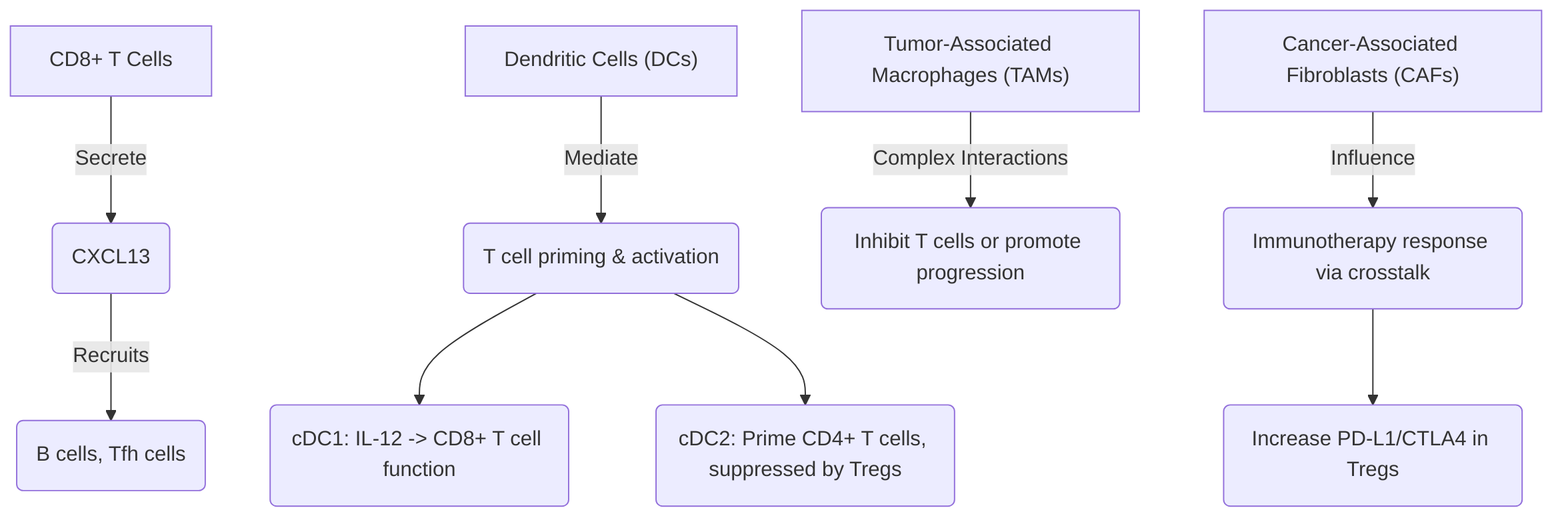
Several studies exemplify the insights gained from single-cell analysis into immune cell interactions. For instance, CD8+ T cells have been observed to secrete CXCL13, facilitating the recruitment of other immune cells such as B cells and T follicular helper cells . Dendritic cells (DCs) are crucial mediators of T cell priming and activation; specifically, cDC1s secrete IL-12 to restore CD8+ T cell effector functions post-PD-1 blockade, while cDC2s are involved in priming CD4+ T cells, often subject to suppression by regulatory T cells (Tregs) . Macrophages, particularly tumor-associated macrophages (TAMs), exhibit complex interactions, with certain TAM subsets potentially inhibiting T cell functions or promoting tumor progression through specific chemokine secretions . Furthermore, stromal cells like cancer-associated fibroblasts (CAFs) can profoundly influence the TME and immunotherapy response. A specific subset of FAP+ CAFs has been identified to drive immunotherapy resistance by increasing PD-L1 and CTLA4 levels in Treg cells via cell crosstalk, underscoring the critical role of these interactions . B cells and natural killer (NK) cells also play potential roles in tumor progression and immunity, with B cell-associated signatures within lymphoid aggregates suggesting their influence on the TIME and potential correlation with patient survival .
The importance of spatial context in understanding these interactions and their impact on immune response and resistance cannot be overstated . Spatial multi-omics technologies, such as PhenoCycler imaging, allow for the analysis of cellular neighborhoods and spatial organization, providing critical insights into cell-cell interactions within the TIME . By integrating spatial multi-omics with computational inference methods, researchers can decipher ligand-receptor interactions, cytokine networks, and the precise impact of spatial proximity on immune cell behavior and their communication with cancer cells .
A significant challenge in dissecting cell-cell interactions lies in inferring causality from mere correlation. While single-cell multi-omics can identify co-localization or co-expression patterns, these correlations do not inherently establish a causal relationship. Spatial data aids in addressing this challenge by providing direct evidence of physical proximity, which is a prerequisite for direct cell-cell contact-dependent interactions or localized paracrine signaling . Observing specific cell types consistently interacting in defined spatial niches allows for more robust hypotheses regarding their functional interdependencies. For example, the enrichment of B cell-associated signatures within lymphoid aggregates, revealed by spatial multi-omics, not only suggests a role for B cells but also provides a spatial context that supports their direct involvement in influencing the TIME and patient survival, moving beyond simple correlative observations . This integration of spatial information with molecular profiles is crucial for building more accurate models of cellular crosstalk and ultimately for establishing causal links in the complex dynamics of the TIME.
3.3 Understanding Immune Checkpoint Blockade Response and Resistance
Single-cell multi-omics is revolutionizing immuno-oncology by offering an unprecedentedly high-resolution understanding of the immune landscape during immunotherapy, thereby enhancing the prediction of patient responses and the identification of resistance mechanisms . This advanced approach facilitates the discovery of specific immune cell subsets and molecular pathways that correlate with treatment efficacy or resistance, providing critical insights into the differential outcomes observed among patients .
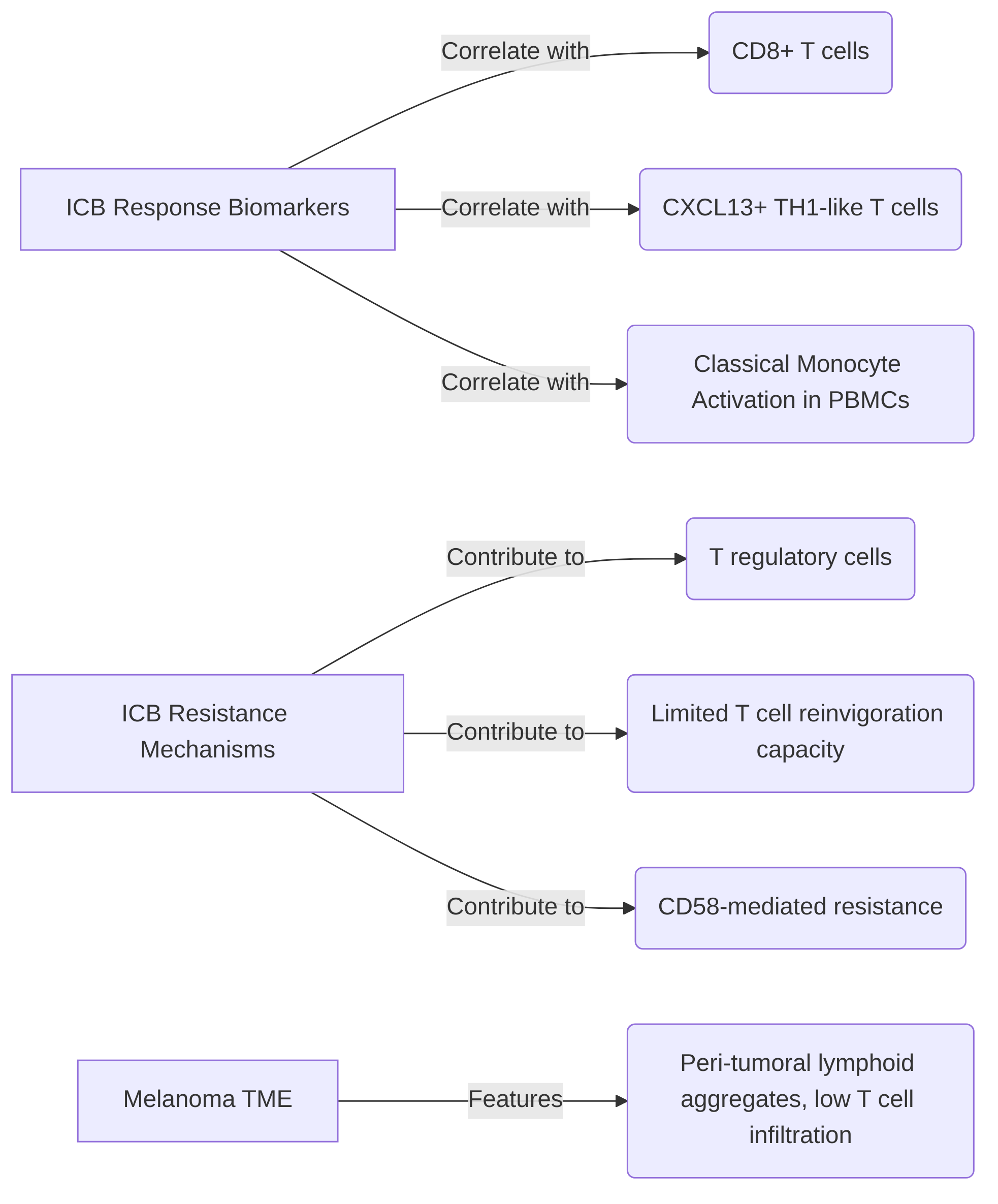
Key findings highlight several predictive biomarkers and resistance mechanisms identified through these technologies. For instance, the presence of CD8+ T cells and specific T helper cell subtypes, such as CXCL13+ TH1-like cells within the tumor microenvironment (TME), has been positively correlated with responsiveness to immune checkpoint blockade (ICB) . Conversely, the sustained presence or elevated levels of T regulatory cells are implicated in contributing to ICB resistance . Beyond tumor-infiltrating lymphocytes (TILs), mass cytometry studies have identified classical monocyte activation in peripheral blood mononuclear cells (PBMCs) as a biomarker for treatment response, indicating that effective immunotherapy can involve organism-wide immune orchestration originating from extratumoral sites like lymph nodes . Monocyte abundance specifically has been noted as a predictor of PD-1 inhibitor treatment response in melanoma patients .
Single-cell multi-omics also critically aids in dissecting resistance mechanisms. For example, some studies indicate that dysfunctional tumor-reactive CD8+ TILs may exhibit limited reinvigoration capacity post-PD-1 blockade, suggesting a need for their cooperation with other immune cells to facilitate the recruitment of new tumor-reactive T cells . Mechanisms of resistance such as CD58-mediated resistance have been identified through the analysis of immune cell profiles . Furthermore, investigations into metastatic melanoma patients have revealed an "immune-striving" TME, characterized by peri-tumoral lymphoid aggregates and low T cell infiltration, as a feature associated with resistance . The emergence of specific melanoma subclones, such as MITF+ SPARCL1+ and CENPF+, post-therapy has also been noted as a potential contributor to acquired resistance, while the enrichment of B cell signatures in lymphoid aggregates correlated with improved survival, suggesting a protective role .
The utility of single-cell multi-omics data in predicting response and identifying resistance mechanisms is substantial, though the robustness and reproducibility of these predictions, along with the biological validation of identified mechanisms, are continuous areas of focus. The ability to analyze T cell receptor (TCR) repertoire diversity and pinpoint peripheral immune signatures contributes significantly to understanding patient responses . While scRNA-seq's feasibility for monitoring clinical responses is increasing , rigorous validation in larger, independent cohorts is essential to confirm the robustness of these single-cell-derived biomarkers. Biological validation of identified resistance mechanisms, such as those related to specific melanoma subclones or immune cell subset dynamics, often involves functional assays and preclinical models to establish causality and explore therapeutic interventions based on these findings .
4. Single-Cell Multi-Omics in Dissecting Tumor Heterogeneity

Single-cell multi-omics technologies have revolutionized the understanding of tumor heterogeneity, offering unprecedented resolution into the complex molecular landscape of cancer. This section explores how these advanced approaches dissect the dynamic transitions of cancer cell plasticity, resolve intratumoral heterogeneity (ITH), facilitate the identification of elusive cancer stem cells (CSCs), and enable the tracing of cancer cell evolution and the emergence of hybrid cellular states. Each of these facets is critical for understanding tumor progression, drug resistance, and ultimately, for advancing precision oncology .
Understanding cancer cell plasticity is paramount, as it describes the dynamic shifts in cellular states that drive tumor heterogeneity and therapeutic resistance . Single-cell multi-omics, by profiling gene expression, chromatin accessibility, and protein levels at a single-cell resolution, provides granular insights into the molecular mechanisms underlying these phenotypic changes . These techniques are instrumental in identifying hybrid cellular states, such as hybrid epithelial/mesenchymal (E/M) cells, which contribute significantly to tumor diversity . Furthermore, they elucidate regulatory mechanisms of processes like epithelial-mesenchymal transition (EMT) and drug resistance, as seen in studies of melanoma subclones emerging post-therapy and the impact of ARID1A deficiency in lung adenocarcinoma . While traditional methods like whole-genome and whole-exome sequencing reveal mutations and copy number variations, single-cell multi-omics integrates these layers, providing a holistic view vital for dissecting complex tumor behavior . Challenges remain in capturing transient plastic states, necessitating longitudinal studies and sophisticated computational integration to reconstruct dynamic trajectories.
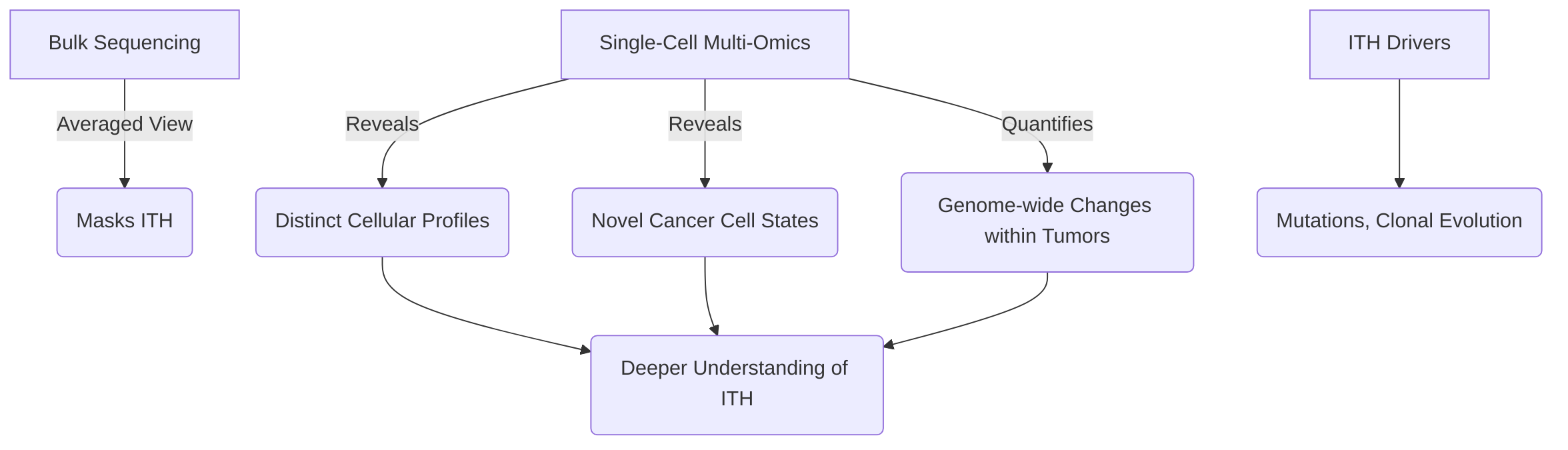
Single-cell multi-omics also provides unparalleled resolution in resolving intratumoral heterogeneity (ITH), a major challenge in cancer driven by mutations and clonal evolution . Unlike bulk sequencing, single-cell approaches reveal distinct cellular profiles, novel subsets, and changes in activation or exhaustion states within tumors, thus accurately quantifying genome-wide changes essential for comprehending tumor heterogeneity . This resolution allows the discovery of novel cancer cell states and resistance mechanisms previously undetectable, such as p-EMT programs in head and neck squamous cell carcinoma (HNSCC) or distinct subpopulations in pancreatic and gastric adenocarcinomas . Single-cell DNA sequencing (scDNA-seq) is particularly effective in identifying genetic heterogeneity, like copy number alterations (CNAs), and integrating multi-omic data enables the reconstruction of tumor evolution trajectories and the identification of molecular events underlying progression and resistance .
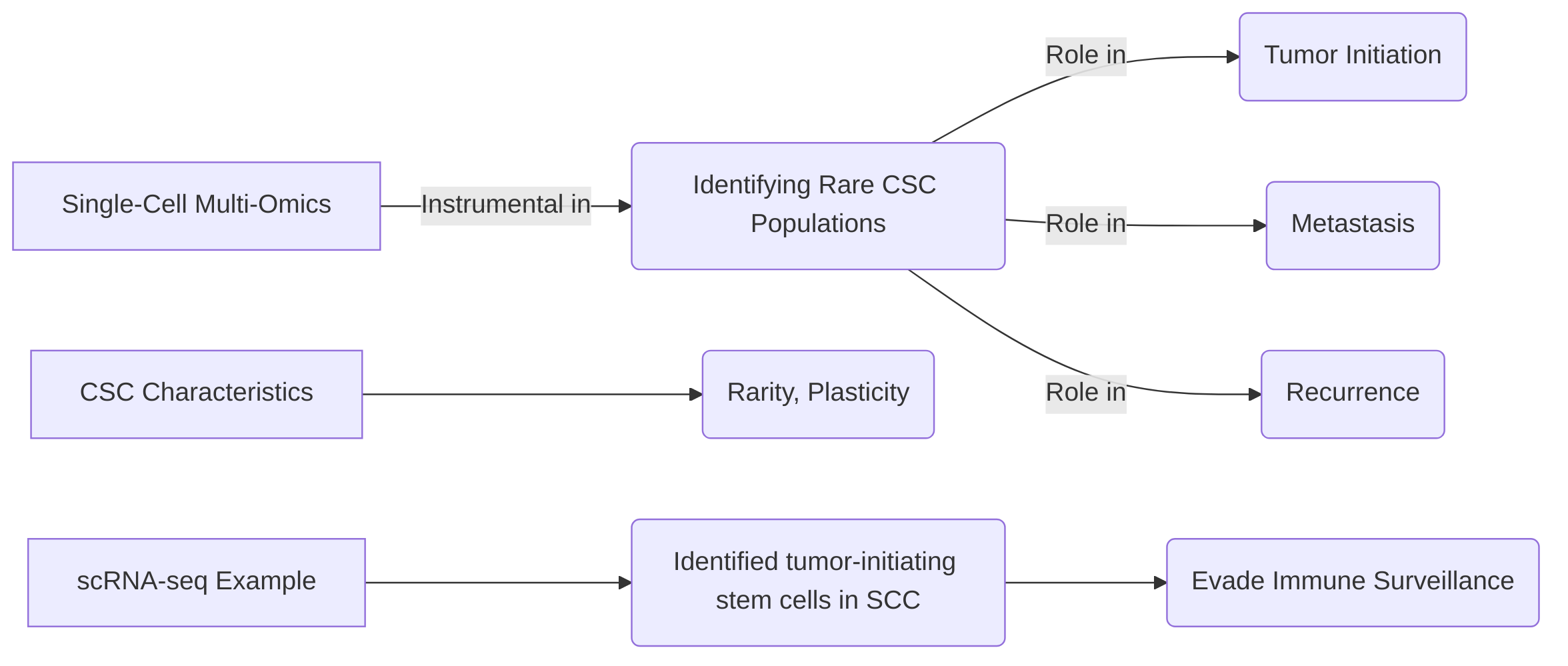
Moreover, these technologies are instrumental in identifying cancer stem cells (CSCs), rare populations often responsible for tumor initiation, metastasis, and recurrence . Single-cell RNA sequencing (scRNA-seq) has successfully identified tumor-initiating stem cells in squamous cell carcinoma that evade immune surveillance . Despite progress, the rarity and plastic nature of CSCs present challenges. Integrating multiple omics layers, such as transcriptomics with ATAC-seq for chromatin accessibility or proteomics, offers a more comprehensive understanding of CSC biology, overcoming the limitations of single-omic approaches by revealing unique regulatory landscapes and functional protein levels.
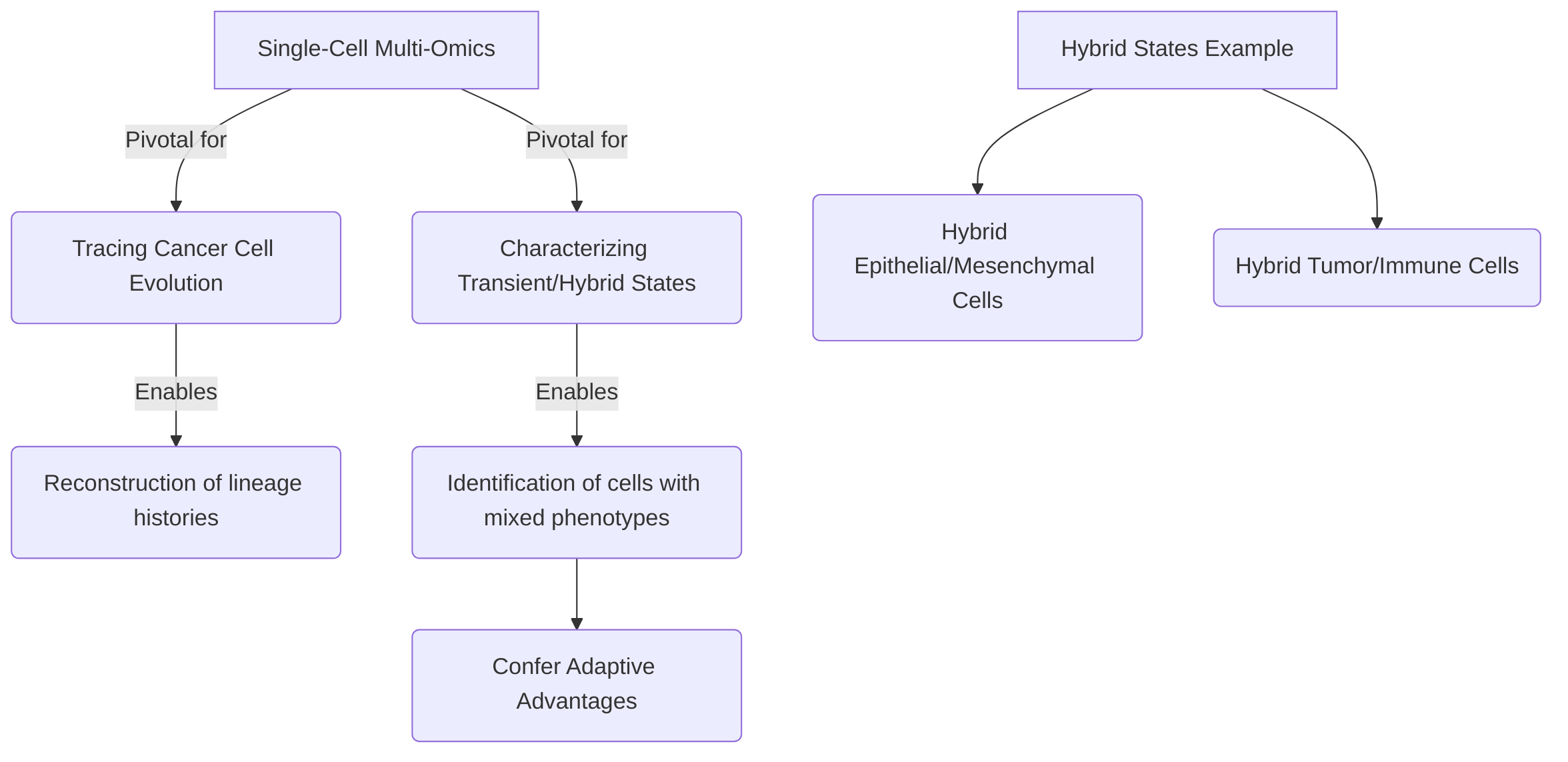
Finally, single-cell multi-omics is pivotal for tracing cancer cell evolution and characterizing transient or hybrid cellular states . These approaches enable the reconstruction of lineage histories and the identification of cells with mixed phenotypes, providing insights into tumor progression and adaptation. Examples include reconstructing lineage trees in myeloproliferative neoplasms (MPNs) and exploring clonal evolution in Multiple Myeloma . Identification of hybrid states, such as epithelial/mesenchymal or tumor/immune cells, underscores their role in conferring adaptive advantages and impacting prognosis, as seen in nasopharyngeal carcinoma . These capabilities offer a more comprehensive understanding of how tumors adapt to selective pressures, including therapy, by mapping changes at the individual cell level and identifying transient, resistant phenotypes. Further development of sophisticated bioinformatics tools will be crucial for fully exploiting the potential of single-cell multi-omics in clinical oncology.
4.1 Understanding Cancer Cell Plasticity
Cancer cell plasticity, defined as the dynamic transitions between distinct cellular states, is a critical driver of tumor heterogeneity and progression, profoundly influencing therapeutic resistance . Single-cell multi-omics approaches offer an unprecedented level of detail into the molecular mechanisms underpinning these phenotypic shifts by comprehensively profiling gene expression, chromatin accessibility, and protein profiles at single-cell resolution .
These advanced techniques enable the identification of hybrid cellular states, such as hybrid epithelial/mesenchymal (E/M) cells, which are prevalent across various tumor types and contribute significantly to overall tumor heterogeneity . For instance, single-cell analysis has revealed the emergence of specific melanoma subclones, including MITF+ SPARCL1+ and CENPF+, following therapeutic intervention, suggesting either treatment-induced plasticity or the selection of pre-existing resistant populations . This capacity to delineate rare, therapy-resistant cell populations is crucial for understanding mechanisms of drug resistance and recurrence.
Furthermore, single-cell multi-omics facilitates the unraveling of regulatory mechanisms underlying processes such as epithelial-mesenchymal transition (EMT), drug resistance, and the acquisition of stem-like properties by cancer cells . By assessing genetic heterogeneity at single-cell resolution, as demonstrated in studies on breast cancer, or by examining cellular states and compositions during tumor progression in conditions like colorectal cancer, these methods provide insights into proliferation, metastasis, and resistance mechanisms, such as those observed in lung adenocarcinoma with ARID1A deficiency .
While whole-genome sequencing (WGS) and whole-exome sequencing (WES) traditionally illuminate mutations and copy number variations, and epigenetic studies (e.g., WGBS, RRBS, ChIP-seq) reveal epigenetic control of gene regulation, single-cell multi-omics integrates these layers of information. Techniques like ATAC-seq, which assesses chromatin accessibility, directly contribute to understanding the dynamic regulatory elements involved in plastic cellular changes . This holistic view is vital for dissecting how complex networks and elements interact to drive tumor behavior, encompassing these plastic transformations.
However, capturing dynamic cellular states with single-cell multi-omics involves trade-offs. The primary challenge lies in preserving the transient nature of plastic states during sample preparation and analysis. While snapshot profiling provides a high-resolution view of existing states, it may not fully capture the temporal dynamics of transitions. Longitudinal single-cell studies or lineage tracing experiments, though technically more demanding, are necessary to track these dynamic shifts over time. Furthermore, the choice of multi-omic platform (e.g., combined transcriptomics and epigenomics vs. transcriptomics and proteomics) can influence the depth of mechanistic insight, with each platform offering distinct advantages in revealing different facets of cellular plasticity. Integration and computational modeling across these diverse data types are therefore essential to fully reconstruct the trajectory of cancer cell plasticity and its implications for tumor evolution and therapeutic resistance.
4.2 Resolving Intratumoral Heterogeneity (ITH)
Single-cell multi-omics technologies have revolutionized the understanding of intratumoral heterogeneity (ITH), a significant challenge in cancer driven by mutations and clonal evolution, by enabling analysis at an unprecedented cellular resolution . Unlike traditional bulk sequencing methods, which provide an averaged molecular profile, single-cell approaches unveil distinct cellular profiles, novel subsets, and changes in activation or exhaustion states within tumors, thus accurately quantifying genome-wide changes critical for comprehending tumor heterogeneity . This granular resolution is instrumental in identifying rare cell populations and their specific molecular features that drive tumor evolution and contribute to treatment failure .
The power of single-cell multi-omics in resolving ITH has led to the discovery of novel cancer cell states and resistance mechanisms previously undetectable by bulk sequencing. For instance, single-cell RNA sequencing (scRNA-seq) has been pivotal in analyzing tumor cells in head and neck squamous cell carcinoma (HNSCC), identifying partial epithelial-to-mesenchymal transition (p-EMT) programs associated with metastasis . In pancreatic ductal adenocarcinoma and gastric adenocarcinoma, scRNA-seq has revealed multiple cancer cell subpopulations, some corresponding to known histopathological subtypes and others representing novel populations with distinct molecular characteristics . Moreover, the emergence of specific melanoma subclones, such as MITF+ SPARCL1+ and CENPF+, was identified after therapy in patients exhibiting resistance to immunotherapy, highlighting the critical role of distinct cancer cell populations in treatment failure . This enhanced resolution has also facilitated the characterization of tumor cell heterogeneity in diffuse large B-cell lymphoma (DLBCL) and the distinct renal cell types and renal tumor cells in renal cell carcinoma (RCC) .
Single-cell multi-omics data is crucial for reconstructing tumor evolution trajectories by providing insights into genetic and transcriptomic changes at the single-cell level. Single-cell DNA sequencing (scDNA-seq) is particularly effective in identifying genetic heterogeneity, such as copy number alterations (CNAs), which are major drivers of ITH . While whole-genome sequencing (WGS) and whole-exome sequencing (WES) have illuminated the contributions of mutations and CNAs to heterogeneity, adapting next-generation sequencing (NGS) technologies to single-cell formats provides the necessary resolution to map these changes to individual cells . By integrating genomic and transcriptomic data, researchers can trace clonal evolution and identify the molecular events underlying tumor progression and therapy resistance. Computational approaches, such as phylogenetic reconstruction algorithms, are then employed to infer lineage relationships and track the emergence and expansion of subclones, thereby delineating the intricate evolutionary paths within a tumor.
4.3 Identifying Cancer Stem Cells (CSCs)
Single-cell multi-omics represents a pivotal advancement in deciphering the intricate biology of cancer stem cells (CSCs), facilitating a more profound understanding of their unique molecular signatures and functional roles within tumors . These sophisticated techniques are instrumental in defining CSC states, thereby illuminating their critical contributions to tumor initiation, metastasis, and recurrence. This capability provides essential insights for both fundamental cancer biology research and the development of targeted therapeutic strategies .
A key success in applying single-cell multi-omics is its capacity to identify rare cancer cell subpopulations that are often responsible for tumor progression and resistance to treatment, a category that frequently includes CSCs . For example, single-cell RNA sequencing (scRNA-seq) has successfully identified tumor-initiating stem cells in squamous cell carcinoma that express CD80 and demonstrate an ability to evade immune surveillance . While the identification of renal cell carcinoma (RCC) stem cells and the potential of WNT and NOTCH inhibitors for their treatment, as well as the correlation between tumor cell stemness in lung adenocarcinoma and prognosis, have been noted, a comprehensive multi-omic characterization of these CSC populations is often not explicitly detailed in general cancer research literature .
Despite these successes, challenges persist in leveraging multi-omics data for CSC isolation and characterization. The inherent rarity of CSCs within heterogeneous tumor masses, coupled with their dynamic and plastic nature, necessitates highly sensitive and comprehensive profiling methods. A critical aspect of applying multi-omics to CSCs involves analyzing the trade-offs of different omics layers in defining CSC properties. While single-cell transcriptomics (scRNA-seq) provides insights into gene expression patterns and stemness markers, integrating additional layers such as single-cell ATAC-seq (for chromatin accessibility), single-cell proteomics, or single-cell epigenomics offers a more holistic view. For instance, chromatin accessibility data can reveal regulatory landscapes unique to CSCs that might not be evident from gene expression alone, while proteomic data can directly quantify functional protein levels. However, the technical complexity, computational challenges in integrating disparate data types, and higher costs associated with comprehensive multi-omics profiling remain considerable hurdles. Each omics layer contributes unique information, but a singular focus on one type may lead to an incomplete understanding of CSC biology, necessitating an integrated approach to fully capture their multifaceted properties.
4.4 Tracing Cancer Cell Evolution and Hybrid States
Single-cell multi-omics technologies have emerged as pivotal tools for dissecting the intricate dynamics of cancer cell evolution and characterizing transient or hybrid cellular states within the tumor microenvironment . By providing a high-resolution view of individual cells, these approaches enable the reconstruction of lineage histories and the identification of cells exhibiting mixed phenotypes, thereby offering profound insights into tumor progression and adaptation .
The capacity of single-cell multi-omics data to trace clonal evolution is exemplified by its application in reconstructing cancer cell lineage trees, such as those observed in myeloproliferative neoplasms (MPNs) based on somatic mutations, and in exploring clonal evolution patterns in Multiple Myeloma . This allows researchers to understand how specific genetic alterations accumulate and shape the evolving tumor landscape.
Beyond clonal evolution, single-cell technologies are instrumental in identifying hybrid cell states, which are critical for understanding cancer cell plasticity and tumor heterogeneity . These hybrid states, such as hybrid epithelial/mesenchymal cells, hybrid tumor/immune cells, and hybrid tumor/endothelial cells, can confer significant adaptive advantages to tumor cells, impacting tumor progression and potentially contributing to therapeutic resistance . A notable example includes the identification of a tumor cell population in nasopharyngeal carcinoma that co-expressed both epithelial and immune markers. This hybrid phenotype was found to correlate with increased tumorigenesis and a poor prognosis, underscoring the functional significance of these transitional states .
The implications of tracing cancer cell evolution and identifying hybrid states are substantial for understanding tumor progression and developing strategies to overcome therapeutic resistance. By dynamically visualizing cancer cell evolution, these technologies offer a more comprehensive understanding of how tumors adapt to selective pressures, including those imposed by treatment . While the foundational principles of tumor heterogeneity and clonal evolution have been illuminated by next-generation sequencing techniques like Whole Genome Sequencing (WGS) and Whole Exome Sequencing (WES) in revealing the impact of multiple mutations or copy number variations , single-cell multi-omics provides an unprecedented resolution to precisely map these changes at the individual cell level, including the emergence of transient, therapy-resistant phenotypes.
Although specific computational approaches for lineage tracing and identifying hybrid states using single-cell multi-omics are broadly mentioned, the digests emphasize the capability of these technologies to reconstruct lineage trajectories and characterize cells with mixed phenotypes . This capability relies on sophisticated bioinformatics tools that interpret complex multi-modal data (e.g., combining genomics, transcriptomics, and epigenomics from the same cell) to infer developmental paths and identify cell populations that do not fit into discrete, well-defined categories. Further research and methodological development in this area will be crucial for fully harnessing the potential of single-cell multi-omics in clinical oncology.
5. Clinical Applications and Future Directions

This section delves into the profound impact of single-cell multi-omics technologies on cancer research, particularly in translating fundamental biological insights into clinical practice and outlining prospective advancements. It begins by exploring how these technologies facilitate biomarker discovery and advance precision medicine, highlighting their utility in identifying predictive markers for treatment response and prognosis, thereby guiding clinical decision-making . Subsequently, the discussion shifts to the pivotal role of single-cell multi-omics in drug target identification and development, emphasizing its ability to dissect cellular heterogeneity and uncover novel therapeutic vulnerabilities within the tumor immune microenvironment (TIME) . Despite these transformative applications, the field faces significant challenges and limitations, encompassing technical hurdles such as data sparsity and loss of spatial information, as well as computational complexities in data integration and analysis, and high associated costs . Finally, the section projects future directions and emerging technologies, advocating for enhanced spatial multi-omics platforms, increased sensitivity in assays, and the integration of artificial intelligence and machine learning to overcome current limitations and realize the full potential of single-cell multi-omics in precision oncology . This holistic view underscores both the current successes and the ongoing efforts required to fully leverage single-cell multi-omics for personalized cancer therapies.
5.1 Biomarker Discovery and Precision Medicine
Single-cell multi-omics technologies have emerged as pivotal tools in biomarker discovery, significantly impacting cancer diagnosis, prognosis, and treatment stratification . These advancements hold substantial potential for guiding clinical decision-making and fostering precision medicine approaches for cancer patients.
The translational impact of these technologies is evident in the identification of specific immune cell subsets or molecular profiles that correlate with clinical outcomes and treatment response. For instance, the abundance of monocytes has been identified as a predictor of response to PD-1 inhibitor treatment in melanoma patients . Similarly, T-cell receptor (TCR) diversity has been recognized as a potential biomarker for personalized therapeutic strategies in precision oncology . Beyond immune cell populations, single-cell RNA sequencing (scRNA-seq) has been instrumental in identifying rare cancer cell subpopulations that drive tumor progression and treatment failure, thereby informing biomarker discovery for prognosis . For example, scRNA-seq aided in the identification of tumor-initiating stem cells in squamous cell carcinoma, with therapeutic implications such as blocking CTLA4-CD80 interaction . Furthermore, single-cell sequencing has illuminated the prognostic roles of stromal cell heterogeneity across various cancers . The density ratio of CD8+/CD3+ T cells has been validated as a prognostic marker in colorectal cancer, underscoring the clinical utility of immune cell quantification within the tumor microenvironment (TME) . Additionally, the enrichment of B cell-associated signatures within lymphoid aggregates has been linked to improved survival, suggesting their potential as biomarkers for predicting immunotherapy response or prognosis .
These technologies also facilitate the development of risk stratification scores and prediction models for treatment response in specific malignancies, such as multiple myeloma and mantle cell lymphoma, enabling individualized treatment approaches . Mass cytometry, for instance, is actively employed in developing high-dimensional cellular biomarkers of treatment response, with studies identifying classical monocyte activation in peripheral blood mononuclear cells (PBMCs) of anti-PD-1 responders . Standardized mass cytometry-based immunophenotyping is also being applied to monitor clinical responses to immunotherapies, and scRNA-seq is anticipated to become increasingly feasible for monitoring immune responses and tumor heterogeneity to guide personalized treatment strategies .
Despite significant progress, the clinical readiness and validation status of many single-cell multi-omics derived biomarkers remain subjects of critical evaluation. While correlations between specific immune microenvironment features and clinical outcomes are being established, a robust immune-based biomarker independent of PD-L1 levels and tumor mutational burden for predicting immunotherapy response is still needed . The primary challenges in translating these biomarkers into clinical practice include the need for extensive prospective validation studies across diverse patient cohorts, standardization of sample processing and data analysis pipelines, and the development of cost-effective and high-throughput assays suitable for routine clinical application. Overcoming these hurdles is crucial to fully harness the potential of single-cell multi-omics in advancing precision oncology.
5.2 Drug Target Identification and Development
Single-cell multi-omics significantly accelerates drug discovery and development by offering an unprecedented resolution into cellular heterogeneity, thereby facilitating the identification of novel therapeutic targets and mechanisms of drug resistance within the tumor microenvironment (TIME) . This approach moves beyond traditional bulk analyses, which often obscure cell-specific vulnerabilities, to uncover precise targets that can be exploited for therapeutic intervention . By integrating multi-level datasets and quantitative tools, single-cell analysis reveals critical interactions that inform the design of new therapies and overcome resistance mechanisms .
One prominent advantage of single-cell resolution lies in its capacity to dissect intra-tumoral heterogeneity, a major challenge in cancer treatment. For instance, single-cell functional proteomics has successfully identified pathway activations associated with drug resistance in melanoma cells, suggesting specific combination therapies to overcome these challenges. The identification of HES6 as a driver of metastasis in uveal melanoma exemplifies a precise, actionable target discovered through single-cell techniques . Furthermore, single-cell multi-omics has revealed the IRE1-XBP1 pathway as a potential therapeutic target in myeloproliferative neoplasms, indicating a specific cell-vulnerability that can be therapeutically addressed .
In specific cancer types, single-cell multi-omics has led to the successful identification of several drug targets. For example, PPIA has been identified as a therapeutic target in Multiple Myeloma . In renal cell carcinoma (RCC), single-cell analyses have illuminated novel therapeutic targets and highlighted the potential of WNT and NOTCH inhibitors for treatment . Beyond cancer cells, targeting components of the TIME has also shown promise, with tumor-associated neutrophils being identified as a potential immunotherapy strategy in liver cancer .
Crucially, single-cell resolution provides unique insights into target selection within the TIME by uncovering the molecular composition of spatial structures, such as lymphoid aggregates, which can inform therapeutic intervention . This approach has highlighted an "immune-striving" tumor microenvironment with low T cell infiltration and identified therapy-induced melanoma subclones (MITF+ SPARCL1+, CENPF+) as potential vulnerabilities or resistance mechanisms to immunotherapy. Such findings are pivotal for developing novel strategies to overcome treatment resistance by specifically targeting these identified subclones or modulating the immune microenvironment . The ability to precisely delineate these cell-specific mechanisms and vulnerabilities at a single-cell level provides a rational foundation for targeted drug development and combination therapies, representing a significant advancement in precision cancer therapy .
5.3 Challenges and Limitations of Single-Cell Multi-Omics
Despite the transformative potential of single-cell multi-omics in dissecting the tumor immune microenvironment (TIME), several significant challenges and limitations persist, impacting its widespread application and the comprehensive understanding of complex biological questions. These limitations can be broadly categorized into technical, computational, and cost-related aspects.
Technically, single-cell multi-omics faces hurdles related to sample preparation, which can introduce perturbations and affect the integrity of transcriptomic and proteomic data, potentially obscuring authentic biological signals . A critical limitation is data sparsity, characterized by a high proportion of zero values in gene expression matrices, which particularly hinders the accurate characterization of rare immune cell populations and their subtle functional states within the TIME . Furthermore, current single-cell techniques often suffer from limited sensitivity, scale, and accuracy, making it challenging to capture the full breadth of cellular heterogeneity and subtle changes in gene expression or protein abundance, thereby impeding a comprehensive understanding of dynamic cellular states in real-time . The loss of crucial spatial information, as most protocols analyze dissociated cells, represents a significant deficiency for TIME research, where cellular interactions and their spatial organization are paramount for immune function and tumor progression. While spatial transcriptomics aims to address this, it often comes with lower resolution, still posing a challenge to precisely map cellular interactions in their native context . Moreover, some single-cell omics approaches, particularly early scRNA-seq techniques, may exclude non-coding genomic regions, potentially overlooking critical regulatory elements and their roles in immune cell differentiation and function .
From a computational perspective, the complexities of data integration, analysis, and interpretation remain substantial . There is a notable lack of standardized computational pipelines for integrating diverse single-cell modalities (e.g., transcriptomics, proteomics, epigenomics, spatial data), leading to inconsistencies across studies and hindering robust comparative analyses . The development of robust bioinformatics tools is crucial for tackling the vast datasets generated by these technologies, especially for understanding emergent behavior beyond reductionist analyses, as emphasized in systems biology approaches . Furthermore, the generalizability of findings can be limited by study design, such as a focus on specific cancer types or patient cohorts, necessitating advanced computational approaches for cross-species translation and broader applicability .
Finally, the high cost associated with single-cell multi-omics technologies continues to be a significant barrier to widespread adoption, particularly in smaller laboratories or resource-limited settings, thereby impeding the scale and breadth of research that can be conducted . Addressing these technical, computational, and cost-related challenges is imperative for realizing the full potential of single-cell multi-omics in uncovering the intricate dynamics of the TIME and translating these insights into effective therapeutic strategies.
5.4 Emerging Technologies and Future Perspectives
The trajectory of single-cell multi-omics in cancer research is marked by rapid technological advancements and sophisticated computational methodologies, promising a deeper understanding of the tumor immune microenvironment (TIME) and paving the way for novel therapeutic strategies . Future advancements are poised to refine existing methodologies, develop innovative techniques, and enhance computational and analytical tools, continuously enriching the single-cell sequencing toolbox .
A critical area of development involves enhanced spatial multi-omics platforms, which are essential for dissecting the complex cellular architecture and interactions within the TIME . For instance, integrating technologies such as CITE-seq with spatial imaging platforms like PhenoCycler has already demonstrated significant power in revealing TIME dynamics and resistance mechanisms to immunotherapy . To address the inherent complexity of spatial data integration and limitations in current spatial resolution, future research should focus on developing AI-driven platforms that leverage principles from statistical physics. These platforms could model spatial interaction networks and cellular communication with unprecedented accuracy, similar to how network theory has elucidated complex systems in other disciplines . This approach would facilitate a transition from descriptive characterizations to functional investigations, enabling rigorous experimental validation of systems-level findings, potentially through immunocompetent animal models and advanced modeling approaches like Agent-Based Modeling (ABM) .
Furthermore, addressing the challenge of data sparsity, particularly for rare immune cell populations, necessitates the development of single-cell multi-omics assays with higher sensitivity. This should be complemented by integrated computational methods for robust imputation, drawing inspiration from historical successes in molecular biology, such as the development of PCR, which dramatically enhanced sensitivity for DNA amplification. Similarly, improvements in single-cell technologies should aim for increased sensitivity, scale, and accuracy .
Future directions also encompass the development of synergistic multi-modal technologies, a deeper understanding of B cell roles in immunity, and improved strategies for T cell infiltration . For example, further exploration into the functional mechanisms of B cells within lymphoid aggregates and investigations into targeted therapies to enhance T cell infiltration into tumors are critical areas for potential therapeutic intervention . The integration of live-cell imaging and protein detection with single-cell multi-omics represents a promising avenue for a more comprehensive understanding of cellular behavior, akin to how advanced imaging techniques from materials science have improved resolution in other fields .
Finally, persistent computational challenges and the high cost of these technologies remain key areas for future development to broaden accessibility and impact . The need for stable, universal, cost-effective, and commercialized methods is paramount, mirroring the impact of high-throughput gene sequencing technologies that revolutionized genomics. The integration of artificial intelligence and machine learning is expected to provide deeper insights into the TME, enabling the transition from descriptive data generation to functionally validated biological discoveries that drive next-generation immunotherapies and personalized care .
6. Conclusion
This systematic review has meticulously explored the transformative impact of single-cell multi-omics technologies on our understanding of the tumor immune microenvironment (TIME), elucidating its critical role in cancer research and the development of precision therapies. The insights gleaned from these advanced techniques have provided an unprecedented resolution into tumor heterogeneity, immune cell diversity, and the intricate cellular interactions within the TIME .
A key finding highlighted in this review is the detailed dissection of intra-tumoral heterogeneity and cancer cell plasticity, which is crucial for identifying diverse cell states and their evolutionary trajectories, ultimately informing the design of targeted cancer therapies . Furthermore, the application of single-cell multi-omics has been instrumental in characterizing various immune cell populations, such as T cells and myeloid cells, and unraveling the complex network of immune cell interactions within the TIME .
Notably, the revelation of specific vulnerabilities within the TIME, particularly in the context of cancer resistance to immunotherapy, represents a significant breakthrough. For instance, single-cell spatial multi-omics has identified an "immune-striving" TIME associated with low T cell infiltration and uncovered specific melanoma subclones that emerge post-therapy, alongside demonstrating the positive association of B cell-enriched lymphoid aggregates with improved patient survival . These advancements are collectively driving progress in biomarker discovery, therapeutic target identification, and the comprehension of treatment response and resistance mechanisms across various cancers . This signifies a critical shift in cancer systems immunology from descriptive analyses towards functional investigations aimed at uncovering emergent behaviors that govern tumor-immune responses .
Despite these profound advancements, several challenges and unanswered questions remain, necessitating sustained research efforts to translate descriptive insights into functional understanding and clinical utility . A critical need exists for functional validation of findings derived from systems immunology studies, demanding rigorous experiments to ascertain the biological and clinical significance of observed cellular states and interactions. This includes leveraging advanced modeling approaches, such as Agent-Based Modeling (ABM), and integrated systems approaches to guide preclinical investigations and inform novel therapeutic designs . A significant gap persists in understanding the precise functional roles of specific immune cell populations, particularly B cells within lymphoid aggregates, and their contributions to anti-tumor immunity. Concurrently, enhancing T cell infiltration and function within tumors remains a primary objective for therapeutic development .
Future research must prioritize technological advancements to improve the sensitivity, scale, and accuracy of single-cell technologies, enabling comprehensive analyses of rare cell populations and subtle molecular changes . Enhancing spatial integration methods through high-resolution and cost-effective spatial multi-omics platforms is crucial for understanding cellular organization and interactions within the TIME . Furthermore, deeper investigation into the molecular mechanisms driving cancer cell plasticity and the identification of novel regulatory pathways, potentially leveraging AI and machine learning for dynamic modeling, is essential for predicting tumor evolution and resistance . Addressing the computational challenges associated with large multi-omics datasets, through the development of robust and user-friendly tools, is also imperative . Finally, reducing the cost and complexity of single-cell multi-omics technologies is vital for broader accessibility and clinical translation, paving the way for novel immunotherapies and precision therapeutics .
6.1 Summary of Key Findings
Single-cell multi-omics has profoundly transformed tumor immunology by providing unprecedented high-resolution insights into the tumor immune microenvironment (TIME) . This technological advancement has enabled a comprehensive understanding of tumor heterogeneity, the remarkable diversity of immune cells, and the intricate interactions between tumor cells and the immune system .
Key contributions include the detailed dissection of intra-tumoral heterogeneity and cancer cell plasticity, revealing diverse cell states and their evolutionary trajectories critical for developing precision cancer therapies . Furthermore, these techniques have elucidated the diversity, states, and functions of various immune cell populations, such as T cells and myeloid cells, and deciphered complex immune cell interactions within the TIME .
A particularly impactful discovery is the revelation of vulnerabilities within the TIME that contribute to cancer resistance to immunotherapy. For instance, single-cell spatial multi-omics, integrating techniques like CITE-seq and PhenoCycler imaging, has characterized an "immune-striving" TIME characterized by low T cell infiltration and identified specific melanoma subclones emerging post-therapy . This approach also highlighted the association of B cell-enriched lymphoid aggregates with improved patient survival, underscoring their potential significance in predicting immunotherapy response .
Overall, the insights gained from single-cell multi-omics are driving significant progress in identifying therapeutic targets, developing robust biomarkers for precision medicine, and comprehending the mechanisms underlying treatment responses and resistance across a broad spectrum of cancers . This marks a crucial transition in cancer systems immunology from mere descriptive characterizations to functional investigations, aiming to uncover emergent behaviors that govern tumor-immune responses and ultimately inform the design of future life-saving treatments .
6.2 Unanswered Questions and Future Research Directions
The burgeoning field of single-cell multi-omics has significantly advanced our understanding of the tumor immune microenvironment (TIME); however, several critical challenges and unanswered questions persist, necessitating focused future research to translate descriptive insights into functional understanding and clinical utility .
A primary unresolved issue lies in moving beyond mere descriptive characterization to achieving functional validation of findings derived from systems immunology studies . This demands requisite experiments to ascertain the biological and clinical significance of observed cellular states and interactions, particularly challenging given the multiscale and multi-organ system nature of tumor-immune dynamics . To address this, future research should leverage advanced modeling approaches, such as Agent-Based Modeling (ABM), to guide preclinical investigations and optimize experimental parameters, especially when animal models fail to adequately predict human responses. Integrated systems approaches that combine diverse datasets and model human biology are crucial for informing novel therapeutic designs .
A significant gap exists in understanding the functional mechanisms of specific immune cell populations, particularly B cells within lymphoid aggregates, and their precise roles in anti-tumor immunity . Concurrently, developing targeted therapies to enhance T cell infiltration and function within tumors remains a critical objective . For instance, future directions should include the development of targeted single-cell multi-omics assays that integrate B cell receptor repertoire sequencing with cytokine profiling and spatial localization to elucidate the specific functional roles of B cell subtypes in anti-tumor immunity. Furthermore, comprehensive investigation into the specific roles of identified melanoma subclones, such as MITF+ SPARCL1+ and CENPF+, in therapy resistance is essential for identifying potential therapeutic targets .
Technological advancements are paramount for addressing current limitations. Future research must prioritize improving the sensitivity, scale, and accuracy of single-cell technologies to enable more comprehensive and precise analyses of rare cell populations and subtle molecular changes . Enhancing spatial integration methods is crucial for understanding cellular organization and interactions within the TIME, requiring the development of advanced spatial multi-omics platforms that are both high-resolution and cost-effective .
Another critical area is the deeper investigation into the molecular mechanisms driving cancer cell plasticity and the identification of novel regulatory pathways . Leveraging AI and machine learning for dynamic modeling of plasticity mechanisms will be vital in predicting tumor evolution and resistance. Addressing the considerable computational challenges associated with the analysis and interpretation of large, complex multi-omics datasets is also imperative. This includes developing more robust, scalable, and user-friendly computational tools and integrated pipelines for dissecting tumor-immune cell crosstalk .
Finally, reducing the cost of single-cell multi-omics technologies is crucial for broader accessibility and clinical translation, advocating for research into developing more accessible, multiplexed platforms suitable for routine clinical adoption . Simplifying experimental procedures, ensuring stable and universal methods, and facilitating commercialization are key steps towards this goal . Integrating single-cell multi-omics with complementary technologies such as gene editing tools and 3D organoid cultures can further expand applications and address current limitations, ultimately paving the way for novel immunotherapies and precision therapeutics .
References
Advancements and applications of single-cell multi-omics techniques in cancer research: Unveiling heterogeneity and paving the way for precision therapeutics - PMC https://pmc.ncbi.nlm.nih.gov/articles/PMC10698529/
Application of Single-Cell Multi-Omics in Dissecting Cancer Cell Plasticity and Tumor Heterogeneity - Frontiers https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2021.757024/full
Applications of Single-Cell Omics in Tumor Immunology - PMC https://pmc.ncbi.nlm.nih.gov/articles/PMC8221107/
Cancer systems immunology - eLife https://elifesciences.org/articles/53839
Applications of Single-Cell Omics to Dissect Tumor Microenvironment - Frontiers https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2020.548719/full
The use of single-cell multi-omics in immuno-oncology - PubMed https://pubmed.ncbi.nlm.nih.gov/35585090/
Single-cell spatial multiomics reveals tumor microenvironment vulnerabilities in cancer resistance to immunotherapy - PubMed https://pubmed.ncbi.nlm.nih.gov/38944836/