0. Recent Advances in CRISPR Base Editing for the Treatment of Inherited Retinal Diseases: A Review
1. Introduction to Inherited Retinal Diseases, Genetic Landscape, and the Need for Advanced Therapies
Inherited Retinal Diseases (IRDs) represent a significant global health burden, affecting approximately 1 in 2000 individuals and serving as a leading cause of blindness, particularly among the working-age population . These conditions are characterized by progressive retinal degeneration, stemming from a complex and heterogeneous genetic landscape involving mutations in approximately 280 to 300 genes, with an estimated 36% of the population carrying at least one IRD-associated mutation . These genetic variations include diverse mutation types such as point mutations (the majority), deletions, insertions, missense, and frameshifting mutations, all impacting genes critical for photoreceptor and retinal pigment epithelium (RPE) function . Clinically, IRDs manifest with varied phenotypes, symptoms, and inheritance patterns, including autosomal dominant, recessive, and X-linked modes . Common IRDs, such as Leber congenital amaurosis (LCA), retinitis pigmentosa (RP), and Stargardt disease, are characterized by progressive vision loss and blindness .
Despite the significant burden, current therapeutic options for IRDs are largely supportive and limited. The sole FDA-approved gene augmentation therapy, Luxturna, targets RPE65-mediated retinitis pigmentosa . However, gene augmentation therapies face several critical limitations, including the requirement for specific RPE65 mutations and a certain threshold of retinal function, restricting patient eligibility. These therapies are not curative, often demonstrate diminishing efficacy over time, and are largely inapplicable to dominant IRDs, which necessitate the inhibition of toxic gene products rather than supplementation . Furthermore, the large coding sequences of many IRD-associated genes (e.g., ABCA4, USH2A, CEP290) exceed the packaging capacity of commonly used adeno-associated viral (AAV) vectors, presenting a significant barrier for traditional gene delivery methods . These limitations highlight a substantial unmet medical need for innovative, more permanent therapeutic solutions that can directly address the underlying genetic defects and halt progressive vision loss.
Gene editing technologies, particularly base editing, offer a promising avenue to overcome these challenges by enabling precise correction of endogenous genetic sequences without relying on large donor templates or risking insertional mutagenesis associated with other viral vectors . Computational methodologies have been crucial in assessing the applicability and scope of base editing for IRDs. A notable approach involves systematically analyzing existing genetic databases such as the Leiden Open Variation Database (LOVD) and patient cohorts from the UK and US with IRDs . This methodology leverages comprehensive variant data to identify pathogenic variants theoretically correctable by base editing, thereby providing a broad overview of targetable mutations and insights into the prevalence of common targetable variants within significant patient populations. The strength of this approach lies in its ability to quantify the therapeutic potential, revealing that a substantial proportion of patients with recessive IRDs possess pathogenic variants amenable to base editing . For instance, approximately 75% of likely pathogenic RPE65 variants are substitutions, rendering them suitable targets for base editing . The underlying assumption is that variants annotated as pathogenic are causative and can be precisely corrected by base editing tools. However, potential biases or limitations may arise if database annotations are incomplete or if the in silico predictions do not fully account for the functional consequences of base editing on non-targetable variants or potential off-target effects. This computational assessment defines the scope of base editing's application, providing critical information for prioritizing research and development efforts toward the most impactful therapeutic targets and informing the potential patient population for future clinical trials. The critical gap that gene therapy, particularly CRISPR-based approaches, aims to address is the direct and permanent correction of the myriad genetic defects underlying IRDs, offering the potential for sustained therapeutic effects and applicability to a broader range of patients, including those with dominant mutations or large gene defects that are intractable with traditional gene augmentation .
1.1 Prevalence and Genetic Landscape of Inherited Retinal Diseases
Inherited Retinal Diseases (IRDs) represent a significant global health burden, affecting approximately 1 in 2000 individuals and serving as a leading cause of blindness among the working-age population in Western countries . The genetic landscape of IRDs is highly complex and heterogeneous, with mutations identified in approximately 280 to 300 genes, highlighting a substantial disease burden given that an estimated 36% of the population carries at least one IRD-associated mutation . These genetic variations encompass a wide range of mutation types, including point mutations (the majority), deletions, insertions, missense, and frameshifting mutations, all of which disrupt genes crucial for photoreceptor and retinal pigment epithelium (RPE) function .
Clinically, IRDs manifest with diverse phenotypes, variable symptoms, and distinct inheritance patterns, including autosomal dominant, recessive, and X-linked modes . Common IRDs include Leber congenital amaurosis (LCA), retinitis pigmentosa (RP), Stargardt disease, and Usher syndrome, all characterized by progressive retinal degeneration leading to severe vision loss and blindness . For instance, LCA, often caused by mutations in genes such as RPE65 and CEP290, results in severe vision impairment from infancy, while autosomal-dominant RP (adRP) linked to rhodopsin (RHO) mutations involves progressive rod photoreceptor degeneration . Mutations affecting key retinoid cycle enzymes can also lead to impaired visual chromophore synthesis or the accumulation of cytotoxic byproducts, contributing to retinal dystrophy .
The current therapeutic landscape for IRDs is markedly limited, underscoring a significant unmet medical need. Existing approaches are primarily supportive, with only one FDA-approved gene augmentation therapy, Luxturna, available for RPE65-mediated retinitis pigmentosa . However, this therapy has several critical limitations. It is not curative and requires specific RPE65 mutations and a certain threshold of retinal function, thereby restricting patient eligibility . Moreover, gene augmentation therapies, while promising, often face challenges such as diminishing efficacy over time, progressive retinal degeneration despite treatment, and their inapplicability to dominant IRDs . The vast genetic heterogeneity, with the need for genotype-specific therapies, makes it difficult to develop a single universal treatment . Furthermore, many IRD-associated genes possess large coding sequences that exceed the packaging capacity of commonly used viral vectors like AAVs, posing a significant hurdle for traditional gene delivery methods . These limitations highlight the urgent need for innovative and more permanent therapeutic solutions, such as gene editing strategies, to address the underlying genetic defects and halt the progressive vision loss associated with IRDs. This establishes the clinical relevance and patient amenability for advanced genetic interventions.
1.2 Computational Assessment of Base Editing Targetability: Mutation Landscape and Patient Population
Computational predictions are crucial for defining the applicability and scope of base editing for inherited retinal diseases (IRDs). While several studies broadly discuss the therapeutic potential of gene editing for IRDs , a focused computational assessment detailing the genetic targetability and patient population amenable to base editing is primarily addressed by one comprehensive study . Other works concentrate on the capabilities of prime editing for various mutation types or specific gene corrections, rather than a broad computational analysis of base editing's targetability across the IRD landscape .
The principal computational methodology utilized for assessing base editing's applicability in IRDs involves analyzing existing genetic databases. A notable study systematically examined data from the Leiden Open Variation Database (LOVD) and patient cohorts from the UK and US with IRDs . This approach leverages comprehensive variant databases to identify pathogenic variants that are theoretically correctable by base editing. The strength of this methodology lies in its ability to provide a broad overview of targetable mutations across a significant patient population, offering insights into the prevalence of common target variants. The underlying assumption is that variants listed as pathogenic in these databases are indeed causative and that base editing tools can precisely correct these specific single-nucleotide substitutions. Potential limitations or biases may arise if database annotations are incomplete or if the functional consequences of base editing on non-targetable variants or off-target effects are not fully accounted for in the in silico predictions.
Quantifying the therapeutic potential of base editing for IRDs, the study focusing on LOVD and patient data revealed that a substantial proportion of patients with recessive IRDs possess pathogenic variants amenable to base editing . This indicates a considerable patient population could potentially benefit from this therapeutic modality, particularly by developing strategies focused on common variants identified through such analyses . For instance, while not a broad computational assessment, a study on Leber Congenital Amaurosis (LCA) caused by mutations in the RPE65 gene noted that approximately 75% of likely pathogenic RPE65 variants are substitutions, rendering them suitable targets for base editing . This highlights that single-nucleotide substitutions, which are the primary targets of base editors, represent a significant portion of the pathogenic mutations in IRDs. The computational assessment serves to define the scope of base editing's application, providing critical information for prioritizing research and development efforts toward the most impactful therapeutic targets and informing the potential patient population for future clinical trials.
2. Preclinical Applications of Gene Editing in IRDs: Base and Prime Editing
This section provides a comprehensive overview of the current research landscape of gene editing for inherited retinal diseases (IRDs), focusing on preclinical applications of base editing and prime editing. We categorize applications by the specific disease or gene targeted, analyzing the reported efficacy and challenges of different approaches, and critically appraise the methodologies used, including the limitations of animal models and the robustness of functional assessment readouts for predicting clinical outcomes .

A direct comparison of delivery strategies, payload limitations, and the range of edit outcomes, such as correction efficiency, mosaicism, and off-target edits, will highlight the translational feasibility of base and prime editing .
Preclinical studies have demonstrated significant progress in utilizing both base and prime editing for IRDs.
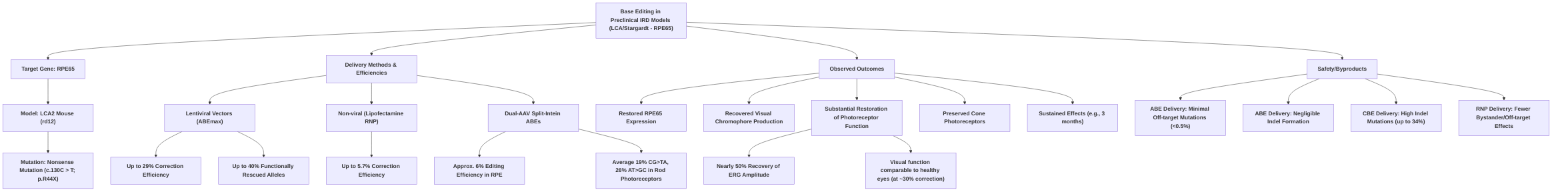
Base editing has shown particular promise in models of Leber Congenital Amaurosis (LCA) and Stargardt disease, primarily targeting the Rpe65 gene . Efficacy has been demonstrated through successful correction of nonsense mutations, restoration of RPE65 expression, visual chromophore production, and significant recovery of photoreceptor function in rd12 mouse models, with visual function comparable to healthy eyes at approximately 30% correction efficiency . Delivery methods for base editing include lentiviral vectors, which have achieved up to 29-40% correction efficiencies and substantial functional rescue , and dual-AAV split-intein systems achieving 6-26% editing in RPE and rod photoreceptors, with sustained effects and negligible off-target editing . Non-viral methods, such as lipofectamine-mediated RNP delivery, while lower in efficiency (1.2-5.7%), have also shown restored Rpe65 expression and improved cone function, with fewer bystander and off-target effects . While ABE-mediated correction has shown minimal off-target mutations and indel events (less than 0.5%) , CBE delivery has led to high indel mutations (up to 34%) .
Prime editing, offering correction of all 12 types of single-nucleotide substitutions, small insertions, and deletions without double-strand breaks , has also demonstrated therapeutic potential in IRD models. In the rd12 mouse model, dual-AAV delivery of prime editor components achieved approximately 6.4% correction efficiency, leading to improved ERG responses and functional rescue, without detectable indels or off-target effects . More advanced dual AAV-split prime editor systems have reached average editing efficiencies of 28% in RPE cells of rd12 mice and remarkably high efficiencies exceeding 76% in retinal cells in a Pde6b mouse model of retinitis pigmentosa, with minimal indel formation and off-target activity, reversing photoreceptor loss and restoring visual function . While prime editing in vivo efficiencies range from 1.71% to over 76%, higher efficiencies are generally observed with optimized dual AAV-split systems .
Both base and prime editing largely utilize murine models (e.g., rd12 mouse model for LCA2 and Pde6b mouse model for RP) that closely mimic human disease phenotypes, enabling assessment of genetic correction and functional rescue via ERG and histological analyses . These models are crucial for evaluating delivery efficiency, tissue tropism, off-target effects, and long-term edit stability, aspects not fully captured by in vitro systems or organoids, which often report higher editing efficiencies (30-50%) .
A critical comparison reveals that base editors are limited to specific transversion mutations (C-to-T/G-to-A or A-to-G/T-to-C), whereas prime editing offers a broader spectrum of correctable mutations . Delivery strategies are a key consideration for translational feasibility. Both base and prime editing face challenges related to payload limitations due to the large size of the CRISPR machinery, often necessitating dual-AAV systems for efficient delivery into retinal cells . While dual-AAV systems have shown promise in achieving efficient cell-type specific delivery to RPE and photoreceptors, optimization of vector tropism and administration routes remains crucial. The range of edit outcomes varies significantly; while some studies report negligible off-target activity for both base and prime editing , others indicate the possibility of underestimated off-targeting rates, particularly for prime editing, without comprehensive genome-wide evaluations . Challenges for both technologies include achieving uniformly high correction efficiency across all targeted cell types in the retina and ensuring long-term safety, especially concerning potential off-target editing, for which consistent and detailed analysis methods are still evolving across preclinical models .
2.1 Base Editing in Preclinical IRD Models
Preclinical studies employing base editing for inherited retinal diseases (IRDs) have demonstrated promising results, particularly in models of Leber Congenital Amaurosis (LCA) and Stargardt disease, primarily targeting the Rpe65 gene. A significant focus has been on the LCA2 mouse model (rd12), which carries a nonsense mutation in the Rpe65 gene (c.130C > T; p.R44X).
Several studies have shown successful correction rates and phenotypic rescue in this model. An Adenine Base Editor (ABEmax) delivered via lentiviral vectors, along with a target-specific guide RNA (gRNA-5), has been instrumental in correcting the nonsense mutation in Rpe65. This approach has achieved correction efficiencies of up to 29% in the rd12 mouse model, leading to the restoration of RPE65 expression, recovered visual chromophore production (11-cis-retinal), and substantial restoration of photoreceptor function and downstream retinal interneurons . Treated mice exhibited nearly 50% recovery of retinal function in ERG recordings, with visual function comparable to healthy eyes . This high level of functional rescue, even with approximately 30% base correction in RPE cells, underscores the translational potential of base editing for IRDs . Furthermore, an optimized ABE lentiviral system achieved up to 40% functionally rescued alleles, protecting cone photoreceptors and counteracting vision deterioration at advanced disease stages .
While lentiviral delivery has shown robust results, non-viral methods such as lipofectamine-mediated delivery of ABE/sgRNA ribonucleoproteins (RNPs) have also been explored, albeit with lower correction efficiencies (up to 5.7% in one study, and 1.8% in juvenile and 1.2% in adult mice in another) . Despite the lower efficiency, these RNP-based approaches still led to restored Rpe65 expression and improved cone function and survival, with fewer bystander edits and off-target effects compared to plasmid-mediated editing . The use of a dual-AAV system with split-intein ABEs in young rd12 mice achieved approximately 6% editing efficiency in the RPE, resulting in increased Rpe65 mRNA, restored RPE65 protein, and enhanced light-induced electrical responses comparable to AAV-RPE65 gene supplementation, with effects sustained for three months and negligible off-target editing . Another study utilizing a split-intein base editor dual-AAV system achieved average editing efficiencies of 19% C•G-to-T•A and 26% A•T-to-G•C in rod photoreceptors .
The range of edit outcomes across studies highlights both the potential and the challenges. While ABE-mediated correction in Rpe65 has consistently shown minimal off-target mutations and indel events (less than 0.5% indel formation and no detected off-target editing at top mutable sites for lentiviral ABE delivery) , a notable limitation was observed with CBE delivery, which led to high indel mutations (up to 34%) . This variation in off-target effects and indel formation between different base editor types (ABE vs. CBE) and delivery methods (viral vs. non-viral) underscores the need for meticulous optimization for each specific application. While some studies broadly discuss CRISPR/Cas9 systems for IRD without specific base editing details , others emphasize the long-lasting restoration of cone functionality and survival attributed to base editing .
Overarching challenges include achieving uniformly high correction efficiency across all cell types in the retina and ensuring long-term safety, particularly concerning off-target editing. Although some studies report negligible off-target activity, the methods for comprehensive off-target analysis are not always detailed or consistent across all preclinical models, representing a specific knowledge gap. The efficiency of delivery, especially with non-viral methods, remains a hurdle for achieving widespread therapeutic effects. Despite these challenges, base editing's ability to provide permanent gene correction, overcoming shortcomings of gene augmentation therapies, strongly supports its continued development for clinical translation in IRDs .
2.2 Prime Editing in Preclinical IRD Models
Prime editing (PE) represents a significant advance in gene editing, offering the capacity to correct a broader spectrum of mutations, including all 12 types of single-nucleotide substitutions, as well as small insertions and deletions, without inducing double-strand breaks (DSBs) or requiring donor DNA repair templates . This mechanism, involving a reverse transcriptase (RT) fused to a Cas9 nickase and a prime editing guide RNA (pegRNA) containing an RT template (RTT), suggests enhanced efficiency and a potentially safer profile compared to traditional CRISPR/Cas9 systems that rely on DSBs and homology-directed repair (HDR) .
Prominent preclinical studies have demonstrated the utility of prime editing in inherited retinal diseases (IRDs), showcasing successful correction rates and significant phenotypic rescue. An early in vivo application in the eye utilized the rd12 mouse model of Leber congenital amaurosis 2 (LCA2), a condition caused by mutations in the RPE65 gene. Dual-AAVs were employed to deliver the prime editor components, achieving approximately 6.4% correction efficiency without detectable indels, unintended substitutions, bystander effects, or off-target effects. This resulted in improved dark-adapted electroretinogram (ERG) responses, reaching up to 67% of wild-type amplitude, indicating substantial functional rescue and the potential for long-lasting visual function restoration .
Further advancements in prime editing delivery and efficiency have been reported. Jang et al. demonstrated subretinal injection of a dual AAV-split prime editor system (AAV-PE2) in the rd12 mouse model, achieving an average editing efficiency of 28% among transduced RPE cells, accompanied by rescue of visual function and an absence of unintended edits . Qin et al. developed a dual AAV-split prime editor system with unconstrained PAM requirements (AAV-PE SpRY) in a Pde6b mouse model of retinitis pigmentosa (RP). This system achieved remarkably high editing efficiencies, exceeding 76% in retinal cells, with minimal indel formation and off-target activity. Functionally, treatment reversed photoreceptor cell loss, restored PDE6β protein levels, and significantly improved visual function, highlighting the translational potential of prime editing for a broader range of IRDs . The Dnmt1 gene was also targeted in adult mouse retina using a dual AAV8 split-PE system, successfully generating a G•A transversion, albeit with a lower efficiency of 1.71% .
When comparing prime editing to base editing, prime editing offers a broader spectrum of correctable mutations, encompassing all 12 types of substitutions, as well as small insertions and deletions. In contrast, base editors are limited to C-to-T/G-to-A or A-to-G/T-to-C transversions . While in vitro experiments in HEK293T cells and organoids have shown high efficiency for prime editing (30-50%) and low undesired editing rates, in vivo efficiency in animal models has been notably lower, necessitating further optimization . The reported correction rates for prime editing in preclinical IRD models range from 1.71% to over 76%, depending on the specific system and target gene, with the higher efficiencies observed in the more optimized dual AAV-split systems . Despite initial concerns, studies like the one in the rd12 mouse model reported no detectable indels, unintended substitutions, bystander effects, or off-target effects at 6.4% efficiency . Similarly, the AAV-PE SpRY system achieved high efficiency with minimal indel formation and off-target activity . However, some studies have shown high levels of unexpected outcomes or unwanted mutations with prime editing, and off-targeting rates may be underestimated without genome-wide evaluation .
The preclinical models predominantly used for prime editing studies in IRDs are murine models, specifically the rd12 mouse model for LCA2 (RPE65 mutation) and the Pde6b mouse model for RP. These models closely mimic the human disease phenotype, allowing for evaluation of both genetic correction and functional rescue through ERG recordings and histological analyses. The choice of these established mouse models facilitates direct comparison with base editing studies, which also extensively utilize similar murine models. The use of these in vivo models is crucial for assessing critical aspects such as delivery efficiency, tissue tropism, off-target effects, and the long-term stability of the edits, which cannot be fully captured in in vitro systems or organoids, where higher editing efficiencies (30-50%) are typically observed . The large size of the PE machinery remains a delivery challenge, though dual AAV-mediated delivery strategies are effectively addressing this limitation and are critical for clinical translation . Optimization of PE design, including PBS length and melting temperature, is also a critical and time-consuming aspect for achieving desired editing outcomes .
3. Overview of CRISPR-Cas Systems and Base Editing Technology
The evolution of CRISPR-Cas systems marks a significant progression in gene editing, transitioning from foundational DNA cleavage tools to highly precise base modification technologies. Initially identified as an adaptive immune mechanism in prokaryotes, the CRISPR/Cas9 system was adapted for mammalian genome engineering due to its RNA-guided targeting specificity . This foundational system utilizes a Cas9 endonuclease directed by a single-guide RNA (sgRNA) to create double-strand breaks (DSBs) at specific genomic loci, contingent on the presence of a protospacer-adjacent motif (PAM) . These DSBs are then repaired by cellular pathways: non-homologous end joining (NHEJ), which is error-prone and often results in insertions or deletions (indels), or homology-directed repair (HDR), which is precise but requires a homologous template and is inefficient in post-mitotic cells such as those found in the retina .

While traditional CRISPR/Cas9 revolutionized gene knockout applications and offered more flexibility than earlier technologies like Zinc Finger Nucleases (ZFNs) and Transcription Activator-Like Effector Nucleases (TALENs), its reliance on DSBs posed significant challenges for therapeutic precision, especially for correcting single-point mutations in non-dividing cells . The high frequency of indels from NHEJ and the low efficiency of HDR in retinal cells limited its applicability for precise gene correction . This limitation, coupled with concerns about off-target activity and potential chromosomal rearrangements, spurred the development of more refined, DSB-independent CRISPR technologies, notably base editing .
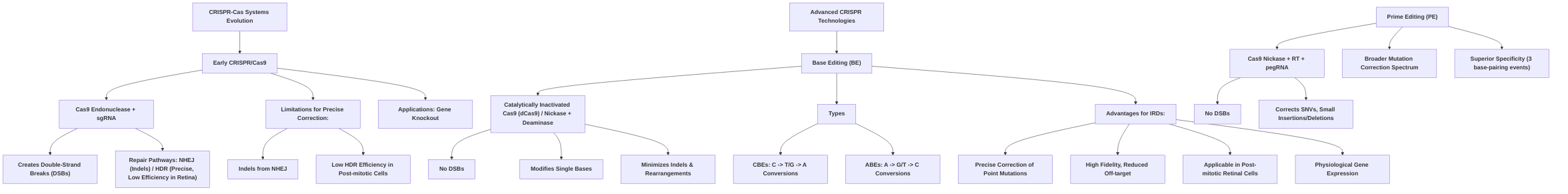
Base editing represents a significant advance, enabling precise nucleotide conversions without inducing DSBs, thereby minimizing unwanted indels and chromosomal rearrangements . This technology typically utilizes a catalytically inactivated Cas9 (dCas9) or a Cas9 nickase fused to a deaminase enzyme, guided by an sgRNA to a specific DNA sequence . The deaminase chemically modifies a target base within an editing window, operating on single-stranded DNA or by creating a single-strand nick, thereby circumventing the error-prone NHEJ pathway prevalent with traditional CRISPR/Cas9 .
Base editors are broadly categorized into Cytosine Base Editors (CBEs) and Adenine Base Editors (ABEs), each designed for specific transitions. CBEs, comprising a Cas9 nickase or dCas9 fused to a cytidine deaminase, convert cytosine (C) to uracil (U), which is subsequently recognized as thymine (T), resulting in a C•G to T•A conversion . These are highly effective for C-to-T or G-to-A transition mutations, which are common in inherited retinal diseases (IRDs) . However, early CBE variants have shown some bystander C deamination and potential for indel formation in non-edited alleles .
ABEs, consisting of a dCas9 fused to an adenine deaminase, convert adenine (A) to inosine (I), which is recognized as guanine (G), leading to an A•T to G•C transition . ABEs are crucial for correcting A-to-G or T-to-C transition mutations, which also represent a significant class of pathogenic variants in IRDs . Preclinical studies, particularly in mouse models of Leber Congenital Amaurosis (LCA), have demonstrated high editing efficiency and precision with ABEs, with minimal indel formation and successful restoration of visual function . Optimized ABE variants, such as ABEmax, have further improved editing efficiency and specificity, significantly reducing off-target edits .
The fundamental advantage of base editing over traditional CRISPR/Cas9 for retinal gene therapy lies in its mechanistic avoidance of DSBs. This circumvents the detrimental effects of NHEJ and allows for efficient, precise correction of point mutations in post-mitotic retinal cells, where HDR is largely inactive . By correcting mutations at their endogenous locus, base editing ensures physiological gene expression, avoiding issues of waning transgene expression common in gene augmentation therapies and addressing dominant IRDs by preventing mutant protein production . While base editing primarily targets transition mutations, advancements have expanded their capability to include select transversion mutations, broadening their therapeutic scope . Ongoing research aims to further optimize delivery methods, enhance specificity, and mitigate potential off-target effects, continually improving the safety and efficacy of base editors for in vivo applications in the retina .
3.1 Basic Principles of CRISPR/Cas9
The Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas9 system, originally discovered as an adaptive immune defense mechanism in bacteria and archaea against viruses and exogenous DNA, has been widely adapted for precise genome engineering in mammalian cells . At its core, the CRISPR/Cas9 system employs a Cas9 endonuclease, which is guided to a specific genomic locus by a single-guide RNA (sgRNA) .
The sgRNA, an artificial fusion of CRISPR RNA (crRNA) and trans-activating crRNA (tracrRNA), is engineered to contain a 20-base pair (bp) sequence complementary to the target DNA sequence . Cas9, typically from Streptococcus pyogenes, possesses two endonuclease domains, HNH and RuvC, which are responsible for cleaving both strands of the DNA . This recognition and subsequent cleavage are contingent on the presence of a protospacer-adjacent motif (PAM) sequence, commonly NGG for S. pyogenes Cas9, located immediately downstream of the target site on the non-complementary DNA strand . Upon successful binding and recognition, Cas9 precisely induces double-strand breaks (DSBs) at the target site .
Following the induction of DSBs, cellular DNA repair pathways are activated to mend the damage. The primary pathways include non-homologous end joining (NHEJ) and homology-directed repair (HDR) . NHEJ is an error-prone pathway that ligates the broken DNA ends directly, often resulting in small insertions or deletions (indels) at the repair site. This characteristic makes NHEJ particularly useful for gene knockout applications, where the introduction of frameshift mutations can inactivate a gene . In contrast, HDR is a high-fidelity repair pathway that utilizes a homologous DNA template to precisely repair the DSB. By providing an exogenous donor DNA template containing the desired genetic modification, HDR can be harnessed for precise gene editing, including the correction of point mutations or the insertion of new genetic material .
The efficiency and specificity of Cas9-mediated gene editing are attributed to the precise RNA-guided targeting mechanism and the requirement for a specific PAM sequence. Compared to earlier genome editing technologies such as Zinc Finger Nucleases (ZFNs) and Transcription Activator-Like Effector Nucleases (TALENs), CRISPR/Cas9 offers significantly greater flexibility and ease of construction, primarily due to the simplicity of reprogramming its specificity by merely changing the sgRNA sequence to target different genetic loci . However, a persistent concern with traditional CRISPR/Cas9 is the potential for off-target activities, where the Cas9 enzyme cleaves DNA at unintended sites due to partial complementarity between the sgRNA and non-target genomic sequences . This challenge has spurred the development of more refined CRISPR-based technologies, such as base editing, to enhance precision and minimize undesired modifications.
3.2 Limitations of Traditional CRISPR/Cas9 for Single-Point Mutations
Traditional CRISPR/Cas9 systems, which rely on inducing double-strand breaks (DSBs), face significant challenges in therapeutic applications, particularly for inherited retinal diseases (IRDs) where high precision and minimal off-target activity are paramount . The core limitation stems from the cellular mechanisms that repair these DSBs: non-homologous end joining (NHEJ) and homology-directed repair (HDR) .
NHEJ, while efficient and highly active in post-mitotic cells such as photoreceptors and retinal pigment epithelium (RPE) cells, is an error-prone pathway that frequently leads to insertions or deletions (indels) . These indels can disrupt gene function, negating the intended correction and potentially leading to deleterious effects, making NHEJ unsuitable for precise single-point mutation correction common in IRDs .
In contrast, HDR offers more precise gene editing by utilizing a homologous DNA template . However, its efficiency is markedly low, especially in non-dividing cells like the photoreceptors and RPE cells that constitute the retina . HDR is predominantly active during the S/G2 phases of the cell cycle, which are largely absent in terminally differentiated retinal cells, thereby significantly limiting its applicability for precise gene correction in this context . For instance, initial attempts to correct RPE65 mutations using CRISPR-Cas9 with HDR demonstrated very low editing efficiency rates .
Beyond efficiency, safety concerns are a critical limitation. DSBs induced by conventional CRISPR machinery can increase the risk of random viral integration during the delivery of CRISPR/Cas components . Furthermore, DSBs pose significant safety risks, including large deletions, chromosomal rearrangements, and the activation of the p53 pathway, which can lead to cell cycle arrest or apoptosis . The "instability and off-target effect of Cas proteins have partially limited its application," as reported by one study, reinforcing the need for more controlled and precise gene editing tools . While HDR has been demonstrated for some IRD gene corrections, its overall low efficiency and the competing error-prone NHEJ pathway in retinal cells make it a less desirable approach for the precise correction of single-point mutations compared to alternative strategies .
3.3 Mechanism of Base Editing and Its Advantages for IRDs
Base editing represents a revolutionary genome editing strategy that enables precise nucleotide conversions without inducing double-strand breaks (DSBs), a significant limitation of traditional CRISPR/Cas9 systems . This technology is particularly advantageous for inherited retinal diseases (IRDs) due to its high fidelity, reduced off-target effects, and applicability in post-mitotic retinal cells, which are crucial for long-term therapeutic outcomes .
The core mechanism of base editing involves a Cas9 component, typically a catalytically inactivated Cas9 (dCas9) or a Cas9 nickase, fused to a deaminase enzyme . The guide RNA (gRNA) directs this complex to a specific DNA sequence, where the deaminase then chemically modifies a target base within a defined editing window. Unlike standard CRISPR/Cas9, which generates DSBs that trigger unpredictable DNA repair pathways often leading to insertions or deletions (indels), base editors operate on single-stranded DNA (ssDNA) or by creating a single-strand nick, thereby preventing DSB formation and minimizing indel rates . This mechanistic avoidance of DSBs is paramount for safety and precision in in vivo therapeutic applications, especially in the delicate retinal environment .
Base editors are primarily categorized into Cytosine Base Editors (CBEs) and Adenine Base Editors (ABEs), each designed for specific nucleotide conversions. Cytosine Base Editors (CBEs) typically consist of a Cas9 nickase or dCas9 fused to a cytidine deaminase, such as rAPOBEC1 . This enzyme deaminates a cytosine (C) to uracil (U) within the target DNA. During DNA replication or repair, the U is recognized as thymine (T), resulting in a C•G to T•A base pair conversion . CBEs are effective for correcting C-to-T or G-to-A transition mutations, which constitute a significant proportion of pathogenic variants in genetic disorders, including IRDs . However, concerns exist regarding their efficiency, product spectrum, and potential for byproduct mutations, such as unintended C deamination or indel formation in non-base-edited alleles, as observed in some preclinical studies .
Adenine Base Editors (ABEs) consist of a dCas9 fused to an adenine deaminase . This enzyme converts adenine (A) to inosine (I), which is subsequently recognized as guanine (G) by cellular repair mechanisms, leading to an A•T to G•C base pair transition . ABEs are crucial for correcting A-to-G or T-to-C transition mutations, which also represent a common class of pathogenic variants in IRDs . Preclinical studies with ABEs in mouse models of Leber Congenital Amaurosis (LCA) (rd12 mice) have demonstrated high editing efficiency and precision, with minimal indel formation and successful restoration of visual function . Optimized ABE variants, such as ABEmax, have further enhanced editing efficiency and specificity, reducing off-target edits below background levels . While both CBEs and ABEs target transition mutations, recent advancements have expanded the capabilities of base editors to include select transversion mutations, broadening their therapeutic applicability .
The key advantages of base editing over traditional CRISPR/Cas9 for IRDs stem from their ability to:
- Avoid Double-Strand Breaks (DSBs): By not inducing DSBs, base editors circumvent the error-prone non-homologous end joining (NHEJ) pathway, which can lead to unpredictable indels and chromosomal rearrangements. This is critical for the safety and precision required for in vivo gene editing in delicate tissues like the retina, particularly in post-mitotic cells where homology-directed repair (HDR) is inefficient .
- Precise Point Mutation Correction: Base editing is specifically designed for single nucleotide variant (SNV) correction, addressing a large proportion of pathogenic mutations causing IRDs, which are often point mutations .
- Endogenous Locus Correction: Base editors correct mutations at their endogenous genomic location, allowing for physiological regulation of gene expression by native promoters, thereby avoiding issues of waning transgene expression seen with gene augmentation therapies and eliminating expression of mutant proteins, which is crucial for dominant IRDs .
- Efficiency in Quiescent Cells: Unlike HDR-dependent methods, base editing does not rely on cell division, making it highly efficient in non-dividing cells such as photoreceptors and retinal pigment epithelial cells, which are the primary targets in many IRDs .
Despite these advantages, challenges remain, including optimizing delivery methods, reducing potential off-target effects, and addressing the specific product spectrum limitations for certain mutation types. Ongoing research focuses on developing improved base editor variants, such as those with expanded editing windows or enhanced specificity, to overcome these limitations and broaden the therapeutic applicability of base editing for IRDs .
3.3.1 Cytosine Base Editors (CBEs)
Cytosine Base Editors (CBEs) represent a significant advancement in CRISPR-based gene editing, specifically designed to introduce precise point mutations without inducing double-strand breaks . Biochemically, CBEs typically consist of a catalytically inactivated Cas9 protein (dCas9) or a Cas9 nickase fused to a cytidine deaminase, such as rAPOBEC1 . This enzymatic component mediates the deamination of a cytosine (C) base within the target DNA sequence to uracil (U) . Subsequently, during DNA replication or repair, this U is recognized as thymine (T), leading to a C•G to T•A base pair conversion .
CBEs are particularly suitable for correcting specific types of point mutations prevalent in inherited retinal diseases (IRDs), primarily those involving C•G to T•A transitions . This mechanism allows for the precise correction of C to T or G to A transition mutations, which account for a substantial portion of pathogenic variants in genetic disorders .
While CBEs offer a promising approach for IRDs, their application requires careful consideration of efficiency, product spectrum, and potential for byproducts. One preclinical study using CBEs in the mouse retina, for instance, reported a 19% C•G-to-T•A editing efficiency in rod photoreceptors . However, this study also highlighted a significant concern: high frequencies of indel mutations were observed in non-base-edited alleles . These indels represent unintended byproducts that could potentially disrupt gene function or lead to new pathologies, underscoring the need for further refinement of CBE specificity and minimization of off-target effects and unwanted editing outcomes. The product spectrum of CBEs is primarily limited to C•G to T•A transitions; they are not inherently designed to correct transversion mutations (e.g., C to A, C to G) without further engineering. Furthermore, the deamination of unintended cytosine bases within the genome and uracil repair-related mutations remain potential off-target effects and sources of byproduct mutations that necessitate thorough evaluation and mitigation strategies in therapeutic applications .
3.3.2 Adenine Base Editors (ABEs)
Adenine Base Editors (ABEs) represent a sophisticated class of genome editing tools engineered to precisely correct A•T-to-G•C point mutations without inducing double-strand breaks . Biochemically, ABEs consist of a catalytically-dead Cas9 (dCas9) fused to an adenine deaminase . This fusion protein facilitates the direct conversion of adenine (A) to inosine (I) on one strand of the DNA, within a target window specified by a guide RNA (gRNA). Inosine (I) is subsequently recognized as guanine (G) by cellular repair mechanisms, leading to the desired A•T to G•C base pair transition . This mechanism allows for the correction of A to G or T to C transition mutations .
ABEs are particularly suitable for correcting a significant proportion of pathogenic point mutations prevalent in inherited retinal diseases (IRDs), given that A•T to G•C transitions are a common class of genetic errors . Unlike Cytosine Base Editors (CBEs), which mediate C•G to T•A transitions, ABEs address a distinct set of genetic alterations, making them complementary rather than competitive in their application scope. While CBEs are effective for C-to-T corrections, ABEs specifically target A-to-G changes, covering different mutational landscapes relevant to IRD pathogenesis . Recent advancements have even expanded the capabilities of base editors, including ABEs, to install select transversion mutations, which typically involve A to G or T to C conversions mediated by adenine deaminases .
The application of ABEs in preclinical models of IRDs has demonstrated promising results. For instance, in mouse models of Leber Congenital Amaurosis (LCA) (rd12 mice) carrying an Rpe65 nonsense mutation, ABEs have shown high efficiency and precision in correcting the mutation . Studies using ABE ribonucleoproteins (RNPs) demonstrated fewer bystander edits and off-target effects compared to plasmid-mediated editing . Furthermore, lentiviral delivery of ABEs achieved high editing efficiencies with minimal indel (insertion/deletion) formation, leading to improved Rpe65 expression and rescued visual function . Optimization efforts, such as the use of an ABEmax variant, have reported high base editing efficiency rates in vitro and in vivo with specific gRNAs, with off-target edits outside the base-editing window being below background levels . A dual-AAV split-ABE system has also demonstrated sustained therapeutic effects comparable to traditional gene supplementation in LCA models, indicating its potential for long-term correction and counteraction of vision deterioration in advanced disease stages .
When assessing the efficiency, product spectrum, and potential for byproducts of ABEs, studies consistently highlight their capacity for precise A-to-G or T-to-C transition mutations . The core advantage of ABEs lies in their ability to achieve specific base conversions without generating double-strand breaks, which are associated with higher risks of indel formation and chromosomal rearrangements. The reported high base editing efficiency, particularly with optimized versions like ABEmax, underscores their therapeutic potential . While the potential for deamination of unintended bases or uracil repair-related mutations (as seen with CBEs) is a general concern for base editors, specific studies on ABEs, particularly with RNP delivery, have indicated a reduced incidence of bystander edits and off-target effects compared to other delivery methods . However, detailed comparative analyses of efficiency and byproduct profiles across various ABE variants and delivery methods, as specifically reported in , are not extensively elaborated upon in the provided digests. Nevertheless, the general consensus suggests that ABEs offer a highly precise and efficient means of correcting specific point mutations, with efforts continually focused on minimizing any unintended genomic alterations.
4. Prime Editing: A Next-Generation Gene Editing Tool for IRDs
Prime editing (PE) represents a significant advancement in gene editing technology, often described as a "search-and-replace" tool due to its enhanced versatility beyond simple single-base substitutions . Unlike traditional CRISPR-Cas9 that induces double-strand breaks (DSBs) or base editing (BE) which is limited to specific transition mutations, PE facilitates the programmable installation of any single-base substitution, small insertion, or small deletion without inducing DSBs or requiring donor DNA repair templates . This expanded capability enables PE to address a broader spectrum of pathogenic genetic variants, potentially up to 89% of known mutations, including all transition and transversion mutations, as well as deletions and insertions, thereby significantly expanding the treatable landscape for inherited retinal diseases (IRDs) .
The core mechanism of prime editing involves a prime editor (PE) protein, which is a fusion of a Cas9 nickase (which nicks only one DNA strand) and a reverse transcriptase (RT), in conjunction with a prime editing guide RNA (pegRNA) . The pegRNA contains both the targeting sequence to guide the Cas9 nickase to the desired genomic location and a reverse transcription template (RTT) that directly encodes the desired edit . After the nickase creates a single-strand break, the RT uses the pegRNA's RTT to synthesize the corrected DNA sequence directly into the target locus . This process mitigates the risks associated with DSBs, such as large deletions or insertions, and eliminates the need for unpredictable cellular DNA repair pathways like homology-directed repair (HDR), which are often inefficient in post-mitotic cells characteristic of retinal tissue . Furthermore, PE demonstrates superior specificity and negligible off-targeting compared to other genome editing technologies due to its requirement for three precise base-pairing events and the direct copying of the desired edit from the RTT sequence, minimizing bystander editing .

The unique therapeutic scope of prime editing for IRDs is particularly evident when contrasting it with base editing. While base editing is highly effective for specific single-base transitions (C-to-T/G-to-A or A-to-G/T-to-C), it cannot correct transversions (e.g., C-to-A, C-to-G) or small insertions and deletions (indels) . Prime editing, however, directly addresses these more complex mutations, offering a solution for a significant portion of IRD-causing variants that are currently intractable with base editing. Preclinical studies in IRD models, such as the rd12 mouse model of Leber Congenital Amaurosis type 2 (LCA2) and Retinitis Pigmentosa (RP), have shown promising results, demonstrating high editing efficiencies, minimal indel formation, and restoration of visual function and photoreceptor survival .
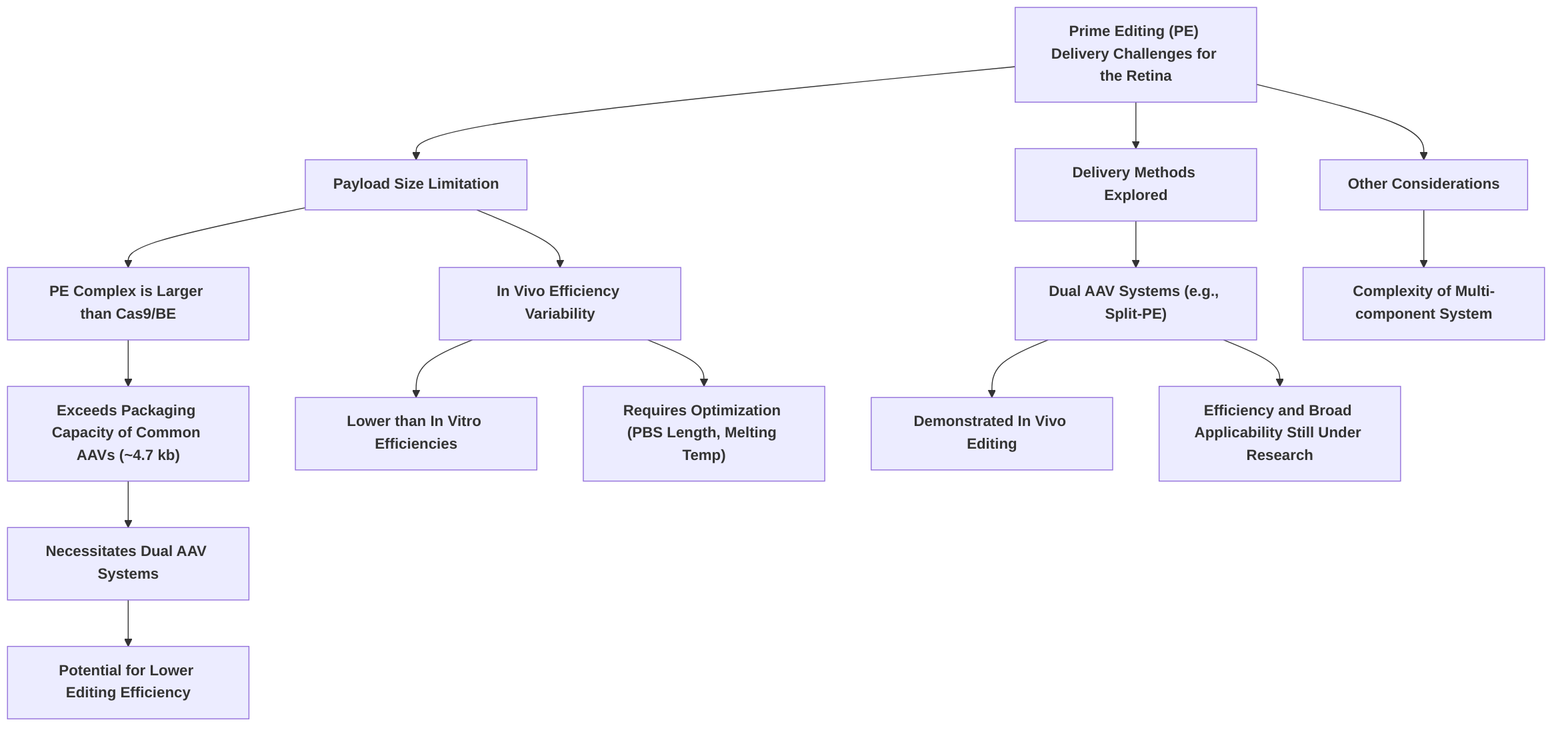
Despite its profound potential, prime editing faces several challenges, particularly concerning its delivery to the retina. The prime editor complex, comprising the Cas9 nickase-RT fusion and the pegRNA, is significantly larger than typical CRISPR-Cas9 or base editing components. This large size poses a substantial hurdle for efficient delivery using conventional adeno-associated virus (AAV) vectors, which have a limited packaging capacity of approximately 4.7 kilobases (kb) . While strategies such as dual AAV systems (e.g., dual AAV8 split-PE systems) have been explored to package the large PE components and have demonstrated in vivo genome editing, their efficiency and broader applicability in the complex retinal environment remain areas of active research . Furthermore, the variability in editing efficiency across different cell types and the complexity of its multi-component system add to the current limitations . Continued research is essential to optimize delivery strategies, improve in vivo efficiency, and thoroughly assess potential off-target effects to fully realize prime editing's transformative potential for IRDs.
5. Challenges and Future Directions
The translation of base editing into widespread clinical application for inherited retinal diseases (IRDs) faces several formidable challenges, necessitating innovative solutions and comprehensive future research. While base editing offers precise, single-nucleotide resolution without inducing double-stranded DNA breaks, concerns persist regarding its off-target effects, efficient and safe delivery, and consistent editing efficiency across diverse contexts . These technical hurdles are compounded by issues of immunogenicity and the complex ethical and regulatory landscape that governs novel gene therapies .
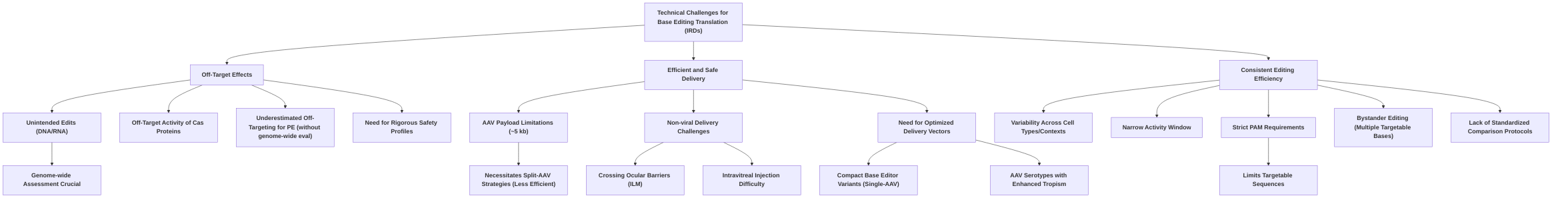
Overarching challenges include the need for rigorous, genome-wide assessment of off-target edits for base and prime editors, moving beyond predicted sites to comprehensive analyses to establish robust safety profiles . The limited payload capacity of AAV vectors, frequently necessitating less efficient split-AAV strategies for the relatively large base and prime editor machinery, significantly impedes efficient delivery to target retinal cells . Non-viral delivery systems, while promising for their cargo capacity and lower immunogenicity, struggle with crossing ocular barriers such as the internal limiting membrane (ILM) following intravitreal injection . Furthermore, the variability in editing efficiency across different cell types and experimental conditions, coupled with the narrow activity window and strict PAM requirements of current base editors, restricts their broad applicability . While base editing demonstrates promising genetic targetability for common IRD variants, its scope is inherently limited to single nucleotide changes, excluding insertions, deletions, and most transversions .
Immunogenicity, primarily from bacterial Cas9 components and viral vectors, remains a critical safety concern, potentially eliciting intraocular immune and inflammatory responses that can compromise therapeutic outcomes . The continuous expression of gene editing components can lead to cytotoxicity and prolonged immune reactions, underscoring the need for transient and tightly controlled expression .
Future research must prioritize holistic, innovative solutions. This includes exploring novel editor designs that boast enhanced precision, broader PAM recognition, and reduced immunogenicity through humanization or the discovery of less immunogenic orthologs. Optimizing delivery vectors is crucial, such as developing compact base editor variants compatible with single-AAV delivery or designing AAV serotypes with enhanced and cell-type-specific tropism for retinal cells. Advancements in non-viral delivery, particularly advanced lipid nanoparticle formulations, are essential to overcome cargo limitations and improve penetration across ocular barriers . Interdisciplinary solutions, integrating insights from nanotechnology, immunology, and biomaterials, can yield novel delivery systems that simultaneously enhance efficiency and reduce immunogenicity. For instance, surface-functionalized nanoparticles could bypass viral vector challenges and improve cellular uptake while minimizing immune responses.
Ethical considerations, while implicitly addressed by the imperative for rigorous safety and efficacy data, demand explicit attention, particularly regarding the distinction between somatic and germline editing. While somatic cell editing for IRDs is ethically more aligned with traditional gene therapy, the broader societal implications of heritable germline modifications necessitate robust public engagement and clear regulatory frameworks . Future research should not only focus on applying base editing in disease-relevant cell types, organoids, and animal models to support clinical potential but also generate comprehensive long-term safety data, including immune responses and genomic stability, which is paramount for regulatory approval . The path to clinical translation, while promising, remains contingent on resolving these complex technical, safety, and ethical challenges through concerted scientific effort and societal dialogue .
5.1 Technical Challenges (Off-target effects, Delivery, Efficiency)
Translating base editing into a routine clinical therapy for inherited retinal diseases (IRDs) necessitates overcoming significant technical hurdles, primarily concerning off-target effects, efficient delivery, and consistent editing efficiency.
One prominent challenge is the potential for off-target activity. While base editors and prime editors generally reduce indel formation compared to conventional Cas9 nucleases, concerns regarding unintended edits persist . Genome- and transcriptome-wide analyses have, in some instances, revealed substantial off-target mutations in DNA and RNA for base editors . The instability and off-target effects of Cas proteins have partially limited their application . Off-target effects can occur due to mismatches between the sgRNA and target DNA, although computational algorithms can predict these sites . For prime editing, off-targeting can be underestimated if analyses solely focus on predicted sites, underscoring the need for comprehensive genome-wide evaluations . While some studies report off-target edits for adenine base editors (ABEs) to be below background levels in vitro , detailed quantitative analysis of in vivo off-target effects often remains underexplored (defect: lack of detailed off-target analysis). Future research should prioritize rigorous, genome-wide assessment of off-target edits across different base editor variants and delivery methods to establish comprehensive safety profiles.
Delivery of gene editing components to target retinal cells efficiently and precisely represents a major bottleneck for in vivo applications . Adeno-associated virus (AAV) vectors are commonly used but are limited by a payload capacity of approximately 5 kb, which can be a significant hurdle for delivering the relatively large base editor (BE) and prime editor (PE) machinery . This often necessitates split-AAV approaches, which, while allowing delivery of larger constructs, can inherently lower editing efficiency . For instance, compact ABEs compatible with single-AAV delivery have been developed, but efficient single-AAV delivery of PE remains challenging . Furthermore, sub-retinal injection, while effective for delivering viral vectors between the retina and RPE, is a technically demanding procedure requiring significant effort to avoid tissue damage and ensure high RPE cell transduction . There is a critical need for a greater understanding of serotype-specific and species-specific differences in retinal tropism and transduction for AAV vectors, which is crucial for translating preclinical data to human clinical trials (defect: insufficient delivery data for specific serotypes and cell types). Non-viral nanoparticle delivery systems offer a promising alternative, potentially overcoming the cargo capacity limitations of AAVs and eliciting lower immunogenicity. However, they face their own challenges, particularly in achieving efficient delivery across ocular barriers like the internal limiting membrane (ILM) following intravitreal injection .
The efficiency of base editing remains variable across different studies and contexts. For example, base editing efficiency has been reported as high as 29% in one study for RPE65-LCA, while a split-PE system in mice showed 1.71% efficiency, highlighting the variability . Prime editing efficiency is particularly variable across different cell types and experimental conditions, requiring time-consuming optimization of design parameters like PBS length and melting temperature . Despite these variations, achieving significant therapeutic effects may not always require 100% editing efficiency; for instance, significant visual function restoration was observed with only 30% base correction in one animal model study . A critical, overarching challenge is the lack of standardized protocols for comparing editing efficiency and safety across different base editing approaches, which impedes robust comparisons and the identification of optimal strategies . The narrow activity window of base editors (typically four to five nucleotides) and the strict PAM sequence requirements further restrict the range of targetable sequences, necessitating the use of base editor variants with broader PAM recognition . Undesired bystander editing can also occur if multiple targetable bases fall within the editing window . While the genetic targetability of base editing for common IRD variants is promising , the specific limitations related to target scope (e.g., exclusion of insertions, deletions, and most transversions) need to be considered .
To address these persistent hurdles, future research directions should investigate adeno-associated virus serotypes with enhanced tropism for specific retinal cell types, such as retinal pigment epithelium (RPE) cells, and explicitly synthesize why certain delivery strategies are more effective or inefficient for specific retinal cell types. This would involve a deeper exploration of how specific AAV capsid modifications or engineering impact transduction efficiency in photoreceptors versus RPE cells. For non-viral delivery, exploring advanced lipid nanoparticle formulations that can efficiently cross ocular barriers, particularly the ILM via intravitreal injection, is crucial. Moreover, interdisciplinary solutions are vital; advancements in biomaterials and nanocarriers could offer innovative solutions for delivering base editing components. Specifically, future studies must develop and validate improved screening methods that better reflect in vivo efficiency and off-target profiles, moving beyond predicted sites to comprehensive genome-wide and transcriptome-wide analyses . Finally, the development of more compact base and prime editor variants, or novel split-delivery strategies with improved reassembly efficiency, is necessary to overcome the current AAV cargo limitations and enhance overall editing outcomes.
5.2 Immunogenicity and Safety Considerations
The safety profile of base editing technologies for inherited retinal diseases (IRDs) hinges critically on minimizing off-target effects and managing immunogenicity, both of which are paramount for long-term therapeutic success. Current base editors offer an advantage over traditional CRISPR/Cas9 systems by avoiding double-stranded DNA breaks (DSBs), which are associated with deleterious rearrangements and oncogenesis . Prime editing further minimizes these risks by avoiding single-strand oligonucleotide donors, thereby reducing DNA toxicity and random integration . While off-target edits outside the base-editing window have been reported to be below background levels in vitro, extensive preclinical in vivo safety data, particularly regarding off-target editing rates for novel base and prime editors in the retinal context, remains limited .
Immunogenicity poses a significant challenge, primarily due to the bacterial origin of CRISPR-Cas9 components and the viral delivery vectors, such as adeno-associated viruses (AAVs). A high percentage of the human population harbors pre-existing antibodies to common Cas9 orthologs like SaCas9 and SpCas9, which can elicit intraocular immune and inflammatory responses following gene editing in vivo . While AAVs are generally considered to have low immunogenicity, they can still induce ocular inflammation and trigger neutralizing antibodies and cytotoxic T-cell responses against the vector, as observed in some clinical trials . For instance, adverse effects reported for EDIT-101 (CRISPR/Cas9 for CEP290-LCA) included retinal tears and hemorrhages, and for sepofarsen (ASO for CEP290-LCA), retinal thinning and cystoid macular edema . The continuous expression of gene editing components can lead to cytotoxicity and immune reactions .
To mitigate these risks, several strategies can be employed. Transient delivery of gene editing components, rather than continuous expression, can reduce prolonged exposure to immunogenic proteins and viral components, thereby minimizing immune responses . Engineering Cas9 variants with reduced immunogenicity, such as by humanizing the protein or identifying less immunogenic orthologs, is another promising avenue. The immune-privileged status of the retina may offer some protection, and the judicious use of immunosuppressive regimens could further ameliorate immune reactions, though this requires careful consideration of systemic side effects . Furthermore, interdisciplinary solutions involving immunology and antibody engineering can inform strategies to block or neutralize pre-existing anti-Cas9 antibodies. Developing non-viral delivery systems, such as lipid nanoparticles or polymeric nanoparticles, offers a holistic solution to simultaneously reduce immunogenicity and enhance cellular uptake through surface functionalization, bypassing the challenges associated with viral vectors and potentially improving safety profiles. Stringent regulatory oversight and comprehensive preclinical and clinical safety data are indispensable for the successful translation of these advanced gene editing therapies to clinical practice.
5.3 Prospects for Clinical Translation and Ethical Considerations
The trajectory of base editing therapies for inherited retinal diseases (IRDs) from preclinical research to clinical application is marked by both significant promise and considerable challenges. Gene therapy, including advanced CRISPR/Cas9 and prime editing, is increasingly recognized as a viable and potentially transformative approach for IRDs, poised to become a mainstay in patient management . The success of Luxturna in gene replacement therapy serves as a foundational example, illustrating the potential for clinical translation within ophthalmology, while recent studies demonstrating encouraging rescue rates in animal models for conditions like LCA further bolster the promise of base editing .
Despite this optimism, several key milestones and challenges must be meticulously addressed to accelerate clinical translation. A paramount concern is the transition from in vitro and early in vivo validation to robust clinical efficacy and safety profiles . Current research emphasizes the need for higher editing efficiency, improved specificity, and enhanced safety, especially concerning potential off-target effects . Achieving sufficient cellular modification within the therapeutic window and ensuring efficient delivery to target retinal cells remain critical technical hurdles . The incremental knowledge gained from ongoing studies and the careful planning and monitoring of research are crucial for simplifying the advancement of CRISPR-based projects . Failures in past trials, such as the ProQR's Brilliance trial for CEP290-mediated LCA, underscore the complexity and the necessity for rigorous preclinical evaluation before clinical implementation .
Ethical considerations are intrinsically linked to the regulatory pathways for gaining approval for novel gene editing therapies. While the provided digests do not extensively detail specific regulatory hurdles or ethical debates such as informed consent, they consistently underscore the imperative for "carefully planned and strictly monitored" research to ensure effectiveness, safety, and specificity before clinical application . The ethical debate surrounding genome editing for inherited diseases primarily revolves around the distinction between somatic cell editing and germline editing. Somatic cell editing, which targets non-reproductive cells and is not heritable, is generally considered less ethically contentious and aligns more closely with traditional gene therapy approaches. Conversely, germline editing, which modifies reproductive cells and results in heritable changes, raises profound ethical concerns regarding unintended consequences for future generations, consent issues for unborn individuals, and the potential for "designer babies" or exacerbating societal inequalities. Although none of the provided digests explicitly discuss germline versus somatic editing, the general emphasis on thorough evaluation of risks and benefits for clinical viability implicitly encompasses these considerations. Regulatory bodies globally are grappling with establishing clear frameworks for gene editing, with stringent requirements for preclinical data on off-target effects, immunogenicity, and long-term efficacy, particularly for novel modalities like base editing.
Future research directions must explicitly integrate the resolution of technical and safety challenges with regulatory and ethical considerations. For instance, advancements in engineering enhanced genome editing tools with ultra-high precision and minimal off-target activity would directly address safety concerns, thereby simplifying regulatory approval processes . Developing highly efficient and safe delivery mechanisms, such as optimized viral vectors or nanotechnology-based carriers, would similarly mitigate risks and facilitate regulatory confidence . Addressing public perception and fostering engagement strategies, informed by transparent ethical discussions, will be critical for the societal acceptance and successful translation of these technologies. This includes open dialogue about the potential benefits for debilitating diseases like IRDs versus the ethical boundaries, especially concerning heritable modifications. Future research should not only focus on applying base editing in disease-relevant cell types, organoids, and animal models to support clinical potential but also on generating comprehensive long-term safety data, which is paramount for regulatory bodies. This includes detailed studies on potential immune responses, genomic stability, and the durability of the therapeutic effect.
Projecting a specific timeline for clinical translation remains challenging due to the multifaceted nature of the identified hurdles. However, given the rapid pace of innovation in gene editing and the significant investment in preclinical validation, we might anticipate initial Phase I/II clinical trials for base editing in IRDs within the next 3-5 years, particularly for specific mutations with strong preclinical data . Broader clinical adoption and regulatory approval for multiple indications could extend to 10-15 years, contingent upon successful outcomes from larger, multi-center trials and the establishment of robust safety profiles.
Ultimately, how will future research addressing technical and safety challenges directly impact regulatory approval pathways and ethical considerations for germline versus somatic editing? Improved precision and reduced off-target activity will undoubtedly ease regulatory scrutiny, particularly for somatic cell therapies, by minimizing unforeseen genomic alterations. For germline editing, demonstrating absolute control over modifications and guaranteeing the absence of unintended consequences across generations would be an unprecedented scientific and ethical feat. Furthermore, rigorous public engagement and education strategies, informed by transparent ethical frameworks, will be pivotal in shaping regulatory receptivity and facilitating the responsible integration of these powerful technologies into clinical practice. This collective effort, encompassing scientific rigor, ethical deliberation, and public dialogue, will determine the ultimate speed and scope of base editing's translation for IRDs.
6. Conclusion and Outlook

This review has comprehensively explored the nascent yet rapidly advancing field of CRISPR base editing for inherited retinal diseases (IRDs), reaffirming its profound potential to revolutionize current therapeutic paradigms. Base editing, as a precise genome editing tool, offers a significant leap forward compared to traditional gene augmentation strategies by enabling permanent gene correction without inducing double-strand breaks (DSBs), thereby mitigating concerns regarding insertional mutagenesis and off-target effects . Preclinical studies have provided compelling proof-of-concept, demonstrating the ability of base editing to correct specific point mutations, restore visual function, and preserve retinal structure in animal models of IRDs, even with partial correction rates of approximately 30% leading to significant visual recovery . This technological advancement holds promise for addressing a substantial proportion of IRD patients, particularly those with common variants and conditions involving larger genes that are challenging for AAV-mediated delivery via conventional gene supplementation .
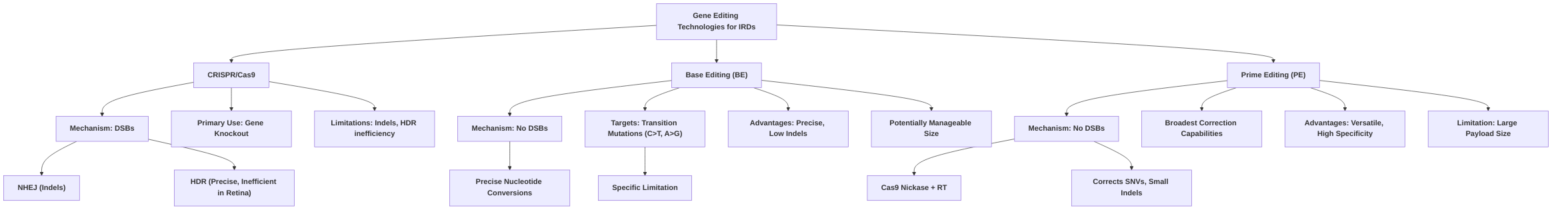
Comparing base editing with prime editing, both represent significant advancements in precise gene correction, moving beyond the limitations of traditional CRISPR-Cas9 by avoiding DSBs . Prime editing, a cutting-edge technique, stands out for its unparalleled versatility, capable of correcting all types of point mutations, small insertions, and deletions without requiring donor DNA templates . While base editing is primarily limited to single-nucleotide conversions (C-to-T/G-to-A and A-to-G/T-to-C), prime editing offers broader mutation correction capabilities . However, prime editing faces a significant hurdle in in vivo delivery due to its larger size, which can exceed the packaging capacity of commonly used viral vectors like AAVs, whereas base editors, particularly optimized variants, may offer more manageable sizes for in vivo applications . Both technologies have demonstrated high efficiency in in vitro settings, but in vivo translational challenges, particularly regarding delivery efficiency and variability, remain key areas for further research .
Looking ahead, the future of CRISPR base editing for IRDs is optimistic yet necessitates continued rigorous research and development to overcome existing limitations and unlock its full therapeutic potential. The field is poised for transformative solutions through synergistic advancements in genome editing tools and sophisticated delivery systems, particularly nanomedicine, as alternatives to AAV vectors for overcoming delivery barriers and ensuring long-term safety and efficacy .
Specific, actionable research priorities for the next 5-10 years include:
- Optimizing Editing Efficiency and Specificity: Developing next-generation base editor variants with enhanced on-target efficiency and reduced off-target activity, particularly in quiescent retinal cells, is crucial. This includes exploring novel Cas protein variants with improved stability and target binding specificity .
- Improving Delivery Strategies: Prioritizing the development of highly efficient and safe in vivo delivery systems for the retina, especially via less invasive routes like intravitreal injection. This involves the continued exploration and optimization of non-viral vectors, such as nanoparticles, which can overcome packaging limitations associated with large base editor components and achieve widespread retinal transduction .
- Broadening Applicability: Expanding the application of base editing to a wider spectrum of IRDs by targeting common disease-causing variants and addressing the genotypic heterogeneity that currently limits therapeutic options for many patients .
- Long-term Safety and Efficacy Studies: Conducting comprehensive long-term studies in larger animal models to thoroughly assess the durability of therapeutic effects, potential immunogenicity, and any unforeseen off-target consequences. This is critical for successful clinical translation and ensuring the sustained benefit and safety for patients .
- Addressing Age-Related Degeneration: Testing base editing strategies in older animal models to determine their efficacy in reversing or halting the progression of established retinal degeneration, analogous to late-stage human disease .

By focusing on these priorities, CRISPR base editing is poised to transition from promising preclinical results to transformative clinical therapies, ultimately improving the quality of life for millions affected by inherited retinal diseases .
References
Genome editing, a superior therapy for inherited retinal diseases - PMC https://pmc.ncbi.nlm.nih.gov/articles/PMC10460145/
The application and progression of CRISPR/Cas9 technology in ophthalmological diseases https://pmc.ncbi.nlm.nih.gov/articles/PMC9998618/
Gene therapy for inherited retinal diseases: exploiting new tools in genome editing and nanotechnology - Frontiers https://www.frontiersin.org/journals/ophthalmology/articles/10.3389/fopht.2023.1270561/full
Therapeutic Gene Editing in Inherited Retinal Disorders - PMC https://pmc.ncbi.nlm.nih.gov/articles/PMC10071418/
In vivo CRISPR base editing rescues cone photoreceptors in a mouse model of early-onset inherited retinal degeneration https://www.nature.com/articles/s41467-024-49340-8
In Vivo Base Editing Corrects Rare Retinal Disease and Restores Sight in Animal Model https://crisprmedicinenews.com/news/in-vivo-base-editing-corrects-rare-retinal-disease-and-restores-sight-in-animal-model/
Novel Therapeutic Approaches for the Treatment of Retinal Degenerative Diseases: Focus on CRISPR/Cas-Based Gene Editing https://pmc.ncbi.nlm.nih.gov/articles/PMC7468381/
Examining the Potential of CRISPR Base Editing for Inherited ... https://www.ajmc.com/view/examining-the-potential-of-crispr-base-editing-for-inherited-retinal-diseases
Prime Editing for Inherited Retinal Diseases - Frontiers https://www.frontiersin.org/journals/genome-editing/articles/10.3389/fgeed.2021.775330/pdf